基于生物信息学方法分析银屑病病理机制中的调节网络
2023-03-02王景乐郎广平韩盈盈
王景乐,徐 曦, 郎广平, 韩盈盈
(1.遵义医科大学 基础医学院, 贵州 遵义 563099;2.遵义医科大学 口腔医学院 口腔疾病研究特色重点实验室,贵州 遵义 563099;3.遵义医科大学 基础药理教育部重点实验室暨特色民族药教育部国际合作联合实验室,贵州 遵义 563099)
银屑病是一种慢性皮肤炎症,临床表现为红斑、脱屑及皮肤增厚[1]。病理特征为角质形成细胞的增殖和免疫细胞的浸润[2]。银屑病分为寻常型银屑病、脓疱型银屑病、红皮病型银屑病以及关节病型银屑病[3],主要影响人的皮肤及关节。各年龄段的人都可能受到影响,患病率具有地理性差异,近年来,银屑病的发病率有所增加[4]。由于银屑病的临床症状出现在皮肤表面,患者身体及心理都有着沉重的负担[5]。银屑病患者会出现各种并发症,如:心血管疾病、精神疾病和代谢综合征;研究表明,银屑病的危险因素包括:肥胖、酗酒和吸烟[6]。目前研究发现IL-23和IL-17是银屑病发病机制中的重要因素,IL-23作为发病机制中的上游,可以刺激Th17细胞的增殖分化,促进下游因子IL-17的产生,进而作用于角质形成细胞,内皮细胞等[7-8]。针对银屑病的治疗目前可采用一些生物疗法,如:TNF-α抑制剂、IL-23抑制剂及IL-17抑制剂等[9]。但是,目前的疗法临床安全性差[10],银屑病仍然无法治愈。因此,寻找关键的靶点及特异性的生物标志物对于银屑病的发病机制研究和临床诊断均有重要意义。
MicroRNA(miRNA)是一种高度保守的小的非编码RNA,可以通过使mRNA翻译受到抑制或通过降解mRNA来调节mRNA的表达[11],作为关键的调节因子,miRNA参与细胞的增殖、分化、凋亡和免疫反应等。如:miR-210在银屑病患者中高表达,促进T细胞分化[12]。因此,miRNA在银屑病发病机制中也有重要的意义[13]。
转录因子(TF)是一种DNA结合蛋白,通过与DNA序列的特定结合位点进行结合,参与转录调控,这是基因表达调控的关键,在基因表达中发挥着重要作用[14-15]。在银屑病中,STAT3已被证实是银屑病发病机制的重要参与者[16]。本研究拟通过公共数据库GEO中2个银屑病数据集的生物信息学分析,筛选出与2个数据集共同差异表达基因互作的蛋白、miRNA、TF以及最有可能调控银屑病病理机制TF-mRNA-miRNA网络。
1 资料与方法
对GEO中的表达谱数据进行差异分析;对差异基因进行富集分析了解差异基因的生物学功能与途径;利用数据库预测差异基因靶向的TF群与miRNA群并构建TF-miRNA-mRNA网络图来识别银屑病相关基因的关键基因、miRNA和TF。
1.1 数据来源 从GEO上下载银屑病表达谱数据集(GSE41662与GSE166388)。GSE41662数据集包含24名银屑病患者的病变皮肤和非病变皮肤样品,该数据集对每名患者进行2次活检,分别进行病变皮肤样品与非病变皮肤样品的收集,将获得的48个皮肤样品在Affymetrix HU133 Plus 2.0微阵列芯片平台进行高通量分析;GSE166388数据集包含4名银屑病患者的银屑病病变皮肤组织和正常健康皮肤组织,将所获得的样本放在Affymetrix HU133 Plus 2.0微阵列芯片平台进行高通量分析。
1.2 差异表达基因(DGEs)筛选 使用GEO2R对GSE41662及GSE166388数据集中的病变组和对照组的基因进行分析筛选,以得到差异表达基因,阈值为P<0.05和log2(FC)≤-2或≥2。
1.3 共同基因的挑选、功能分析和通路分析 利用Venny2.1将2个数据集的差异基因进行合并找出共同的差异基因;使用R(3.6.3版本)中的org.Hs.eg.db包对共同差异基因进行ID转换;以P<0.05为阈值,利用clusterProfiler包对共同差异基因进行GO和KEGG富集分析后,以分析共同差异基因参与的生物学过程(BP)、细胞成分(CC)、分子功能(MF)情况以及KEGG富集的通路结果,最后利用ggplot2包将结果进行可视化。
1.4 共同差异基因的PPI网络构建及表达分析 本研究利用STRING构建共同差异基因的PPI网络图后,使用Cytoscape中CytoHubba插件对该网络进行关键基因的筛选,根据MCC方法评分挑选,MCODE插件对基因进行构建聚类功能模块,参数设置为degree cutoff=2、node score cutoff=0.2、k-core=2、and max depth=100。
1.5 目标miRNA、转录因子预测及网络构建 利用Targetscan和miRWalk对共同差异基因的靶向miRNA进行预测后,使用Cytoscape构建miRNA-mRNA网络图;使用TRRUST预测共同差异基因的转录因子(TF),Cytoscape构建TF-mRNA网络图。最后构建TF-mRNA-miRNA网络图。
1.6 统计学分析 使用GEO2R、R(3.63版本)ClusterProfiler包、ggplot2包、org.Hs.eg.db包以及Cytoscape软件对公共数据集进行分析。P<0.05时差异具有统计学意义。
2 结果
2.1 差异基因的筛选 为了找出两组数据集中银屑病患者组与正常人组的差异表达基因,利用GEO2R对各样本进行差异分析。经过差异分析可视化后发现,GSE41662与GSE166388两个数据集的各自样本的中位数及上四分位数基本一致(见图1A、B),这表明样本间归一化程度好。GSE41662与GSE166388中的病变组与对照组基因表达谱存在明显差异(见图1C、D)。以P<0.05为阈值制做出的火山图,其中红色为表达上调基因,蓝色为表达下调基因(见图1E、F)。从GSE166388数据集中获得206个差异基因(66个下调和140个上调基因),其中高表达前10位的基因为SERPINB4、DEFB4B、S100A7A、CMPK2、IFI44L、S100A12、IFI6、S100A9、ISG15和IFIT1,低表达前10位的基因POSTN、MIR675、CMAHP、HS3ST6、BTC、C3orf52、NELL2、KRT77、SLC25A27和MSH5-SAPCD1;GSE41662数据集中获得205个差异基因(44个下调和161个上调基因),其中高表达前10位的基因是S100A7A、SPRR2C、DEFB4B、SERPINB3、TCN1、CXCL8、PI3、GDA、IGFL1和IL36G,低表达前10位的基因是THRSP、SLC14A1、RORC、GPD1、WIF1、TPPP、THRSP、SCGB2A1、WDR72和MYO3A。

A、B:2个数据集差异分析可视化的箱线图;C、D:2个数据集差异分析可视化的UMAP图;E、F:2个数据集差异分析可视化的火山图。
2.2 差异表达基因的GO与KEGG通路富集分析 为了找出2个数据集中的差异基因所参与的功能,首先利用Venny2.1将GSE166388与GSE41662的差异基因合并找出共同的差异基因,得到57个差异基因,包括EPSTI1、OAS1、KYNU、STAT1、IL36G、CD24、RSAD2、S100A7A、PI3、OASL等(见图2A);利用R(3.6.3版本)中的org.Hs.eg.db包对基因ID进行转换,最后利用clusterProfiler包对57个共同差异基因进行富集分析。GO富集结果表明,57个差异基因主要富集在角质化、特定颗粒腔、编码角质化包膜和Ⅰ型干扰素信号通路等过程,其中在BP上富集在角质化、嗜中性白细胞活化及Ⅰ型干扰素信号通路3个途径;在CC上富集在分泌颗粒腔、编码角质化包膜和特定颗粒腔3个途径;在MF上富集在趋化因子活性、细胞因子受体结合和趋化因子受体结合3个途径;在KEGG上富集在丙型肝炎、IL-17信号通路及甲型流感途径(见图2B、C)。图2D是对富集分析得到的结果进行可视化形成的网络图,结果与图2B,图2C相同。
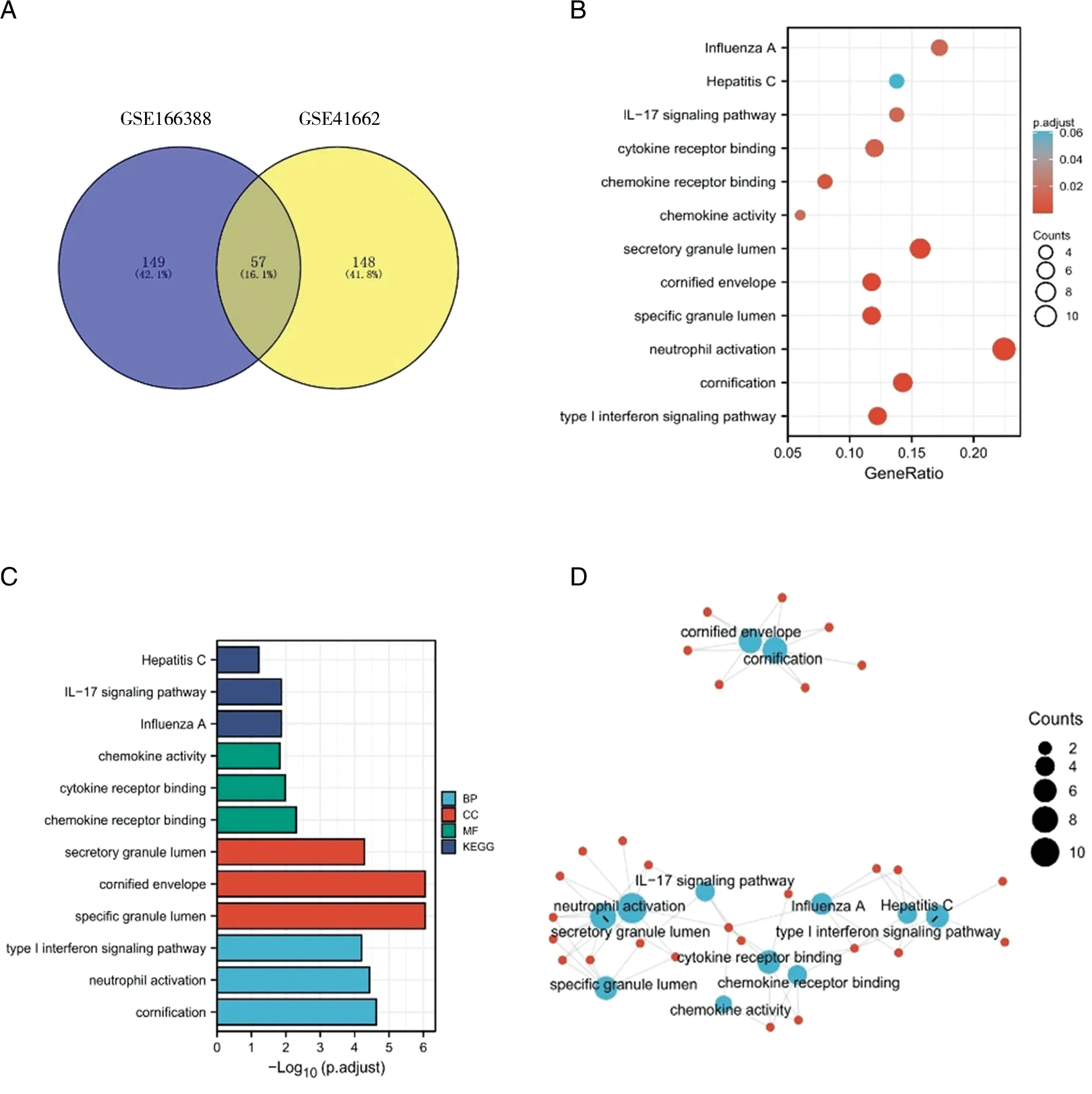
A:GSE166388与GSE41662数据集差异基因合并的维恩图;B:GO与KEGG富集分析可视化气泡图;C:GO与KEGG富集分析可视化柱状图;D:GO与KEGG富集分析可视化网络图。
2.3 PPI网络构建与Hub基因鉴定 为了解析57个差异基因与对应的蛋白质的相互作用关系,首先,利用STRING数据库对57个基因构建出由54个点和87条边构成的PPI网络图。其次,利用Cytoscape对网络图进行美化(见图3A)。最后,使用Cytoscape中的CytoHubba插件筛选top20hub基因(见图3B),其中, MCC法评分最高的基因是STAT1,该基因可以参与到免疫反应当中,该结果表明STAT1很可能参与到银屑病的发病机制中;评分第二的是RSAD2、OAS1,其中RSAD2可以编码抗病毒蛋白,参与到抗病毒反应与免疫反应当中;评分第三的是EPSTI1、OASL、SAMD9和OAS2,其中OAS2可以编码2-5A合成酶家族,参与到免疫反应当中;MCODE插件构建功能模块,在建立分数>2时,获得5个模块,前3个构成分别为:7个点21条边(见图3C)、5个点10条边(见图3D)、4个点6条边(见图3E)。
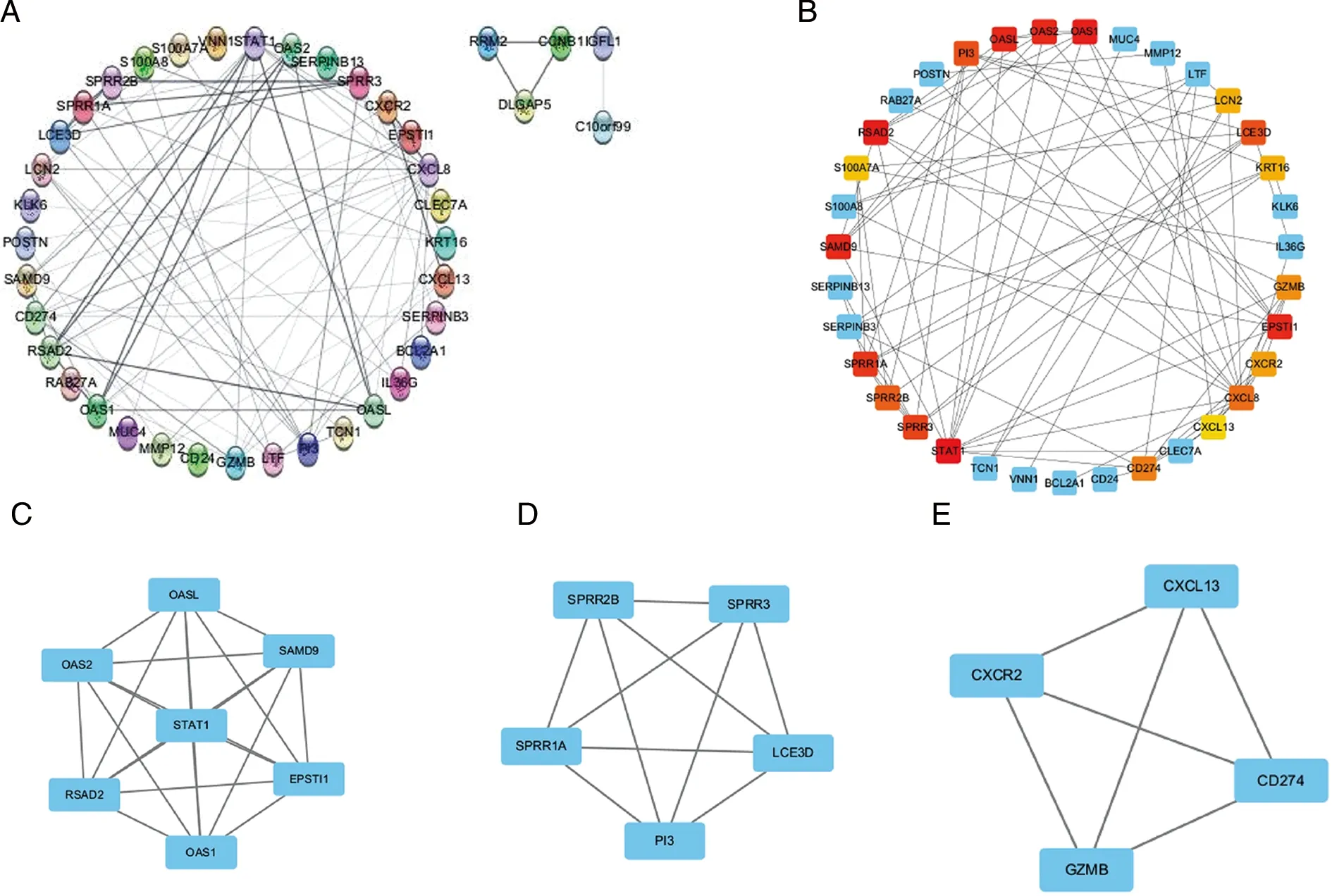
A:STRING构建的差异基因PPI网络图(54个节点,86条边);B:利用Cytoscape筛选出的前20Hub基因(颜色越深,评分越高);C-E:Cytoscape中的MCODE插件构建的3个模块。
2.4 miRNA与TF预测及网络构建 为了解与分析出的差异基因相关的转录因子和miRNA,首先分别利用miRWalk与Targetscan预测前20个关键基因的miRNA,其次利用TRRUST预测前20个关键基因的转录因子,得到9个转录因子分别是LF3、NFE2L2、RELA、STAT3、JUN、STAT6、EP300、FOS和NFKB1,其中RELA调控CXCL8、CXCR2、LCN2、STAT1 4个基因,调控基因数排名第一,这说明RELA参与到银屑病的可能性更大。最后,利用cytoscape构建miRNA-mRNA网络(见图4A),如:miRNA-3064-5p调控STAT1;mRNA-TF网络(见图4B),如:STAT1-STAT3,合并构建TF-mRNA-miRNA网络(见图4C),如:RELA-STAT1-miRNA-3064-5p。RELA与STAT1都可以参与到炎症当中,而miRNA-3064-5p也与细胞增殖分化有关,这提示RELA-STAT1-miRNA-3064-5p可能参与到银屑病机制当中。
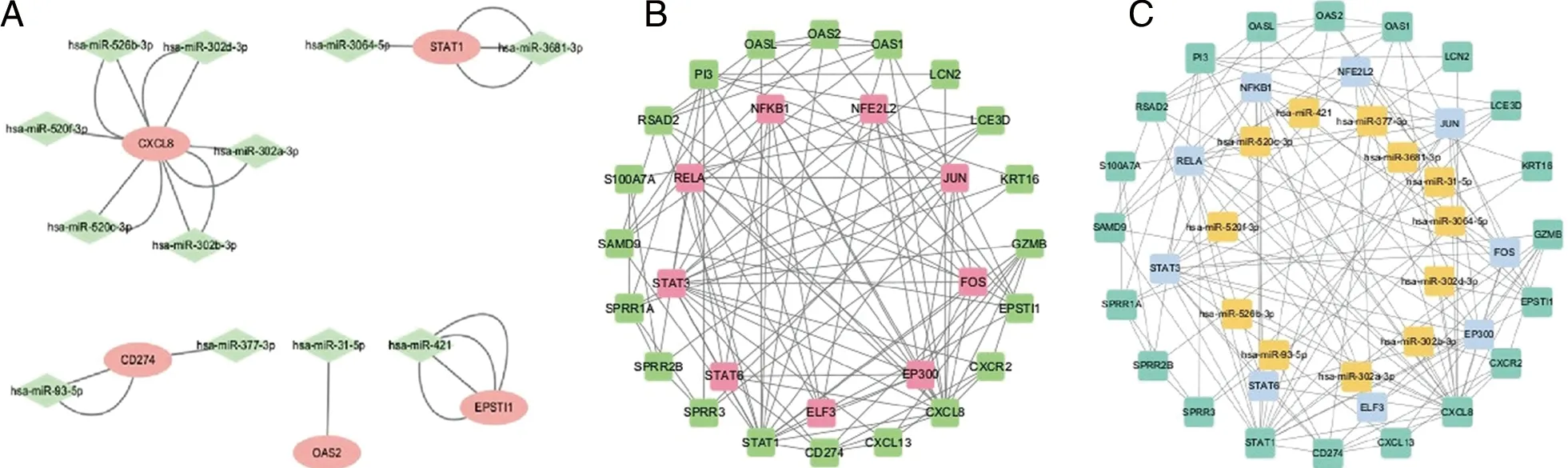
A:靶向共同差异基因的miRNA与共同差异基因构建的网络图,粉色为共同差异基因,绿色为miRNA;B:共同差异基因预测出的转录因子与共同差异基因构建的网络图,绿色为共同差异基因,粉色为TF;C:预测的miRNA、转录因子、共同差异基因共同构建的网络图,绿色为共同差异基因,蓝色为TF,黄色为miRNA。
3 讨论
皮肤是人体的第一道防线,其免疫细胞如树突状细胞、巨噬细胞、朗格汉斯细胞等在免疫反应中起着至关重要的作用。银屑病是一种主要由T细胞介导慢性炎症性皮肤病,其特点在于上皮角质细胞的过度增殖及异常分化[17-19]。在这项研究中,我们通过分析数据集中的正常皮肤组织和银屑病病变组织的基因表达谱,从GSE166388数据集中获得206个差异基因,GSE41662数据集中获得205个差异基因,将两个数据集的差异基因进行合并后得到57个共同的差异基因。在对57个差异基因进行富集分析后,GO富集结果表明,BP中富集基因最多的是嗜中性白细胞激活途径;CC中富集基因最多的是分泌颗粒腔途径;MF中富集基因最多的是细胞因子受体结合途径,这表明在银屑病过程中,细胞因子由特定细胞(如树突状细胞)分泌后作用于靶细胞(如T细胞)进而参与银屑病的发生[19-20]。KEGG富集分析显示57个差异基因富集在丙型肝炎、IL-17信号通路、甲型流感和趋化因子信号通路等通路中。已有的研究表明:IL-17是一种促炎因子,参与机体的防御,屏障组织的修复,炎症反应等过程,在银屑病中发挥重要作用[21-22]。趋化因子是机体免疫系统的一部分,可控制免疫细胞的迁移,将免疫细胞募集到病变部位[23]如CCL20/CCR6轴,在银屑病中角质形成细胞受到刺激后分泌CCL20募集T细胞到病变皮肤发挥作用[24]。根据既往研究,在银屑病的发病过程中,细胞因子如IL-17A扮演着至关重要的角色,而本研究的富集结果表明,57个差异基因大部分富集在细胞因子相关途径,但在KEGG富集分析中,有基因富集在流感与肝炎中,这可能是因为流感与肝炎都会影响机体的免疫反应与炎症反应,所以存在与银屑病相同的通路和相关基因或因子,但是肝炎或流感与银屑病并发的机制尚不清楚[25],而本研究通过富集分析也证实了肝炎或流感与银屑病相关的观点。总之,经过以上讨论,本研究的富集结果与既往研究结果一致,因此,细胞因子受体或IL-17信号通路等途径可参与到银屑病的发病过程,而肝炎或流感与银屑病相关。
STAT1是IFN-α/β信号传导通路的关键基因,参与宿主的防御[26],编码的蛋白质是STAT蛋白家族的成员,在免疫反应中起着重要作用,主要是通过JAK-STAT 通路发挥作用,当细胞因子受体与配体结合后,激活JAK激酶(JAKs),JAKs进而激活STAT1[27]。目前,STAT1已被证实参与到银屑病病理过程中,调控银屑病标志物K17的表达,如:IL-17A可以通过激活STAT1使K17表达上调进而参与银屑病的发病机制[28]。此外,IL-27可以通过激活STAT1产生趋化因子或IL-18结合蛋白参与到银屑病发病机制中[29-30]。本研究通过利用Cytoscape对由57个基因构建出的PPI网络进行分析后,STAT1是MCC法评分最高的关键基因,表明STAT1参与并在银屑病中发挥重要作用。miRNA-3064-5p目前在银屑病中尚无研究,其研究主要集中在癌症方面,研究表明miRNA-3064可以抑制细胞的增殖分化,miRNA-3064可以通过靶向hTERT来控制卵巢癌细胞的增殖[31-32]。作为miRNA-3604家族的一员,miRNA-3064-5p可通过长链非编码RNA的调控对细胞增殖分化发挥作用,如:长链非编码RNA AGAP2-AS1可通过调节miR-3064-5p的表达促进宫颈癌细胞的增殖、MALAT1可调节miR-3064-5p的表达促进乳腺癌细胞的增殖和迁移[33-34]。本研究在对调控前20个关键基因的miRNA预测后,发现miRNA-3064-5p能够靶向STAT1。目前,STAT1与miRNA-3064-5p之间的作用尚无报道,但由于miRNA-3064-5p可以调控细胞的增殖和分化,我们可以推测miRNA-3064-5p有可能靶向STAT1参与到银屑病的发病过程中。
RELA是NF-κB的家族成员之一,是NF-κB通路中的关键成员,又称P65,参与调节炎症、细胞增殖和凋亡[35-36]。RELA可抑制细胞凋亡控制细胞增殖的速度,诱导促炎因子的产生,当RELA与FOXO3结合形成复合物以后可抑制促炎信号通路的传导和功能,此外,RELA在肺炎等炎症性疾病中至关重要,疾病期间的保肝作用依赖RELA的诱导[35,37-38]。而RELA在银屑病中研究有限,研究发现银屑病患者皮肤中的磷酸化RELA升高并激活NF-κB通路,继而使细胞因子如:TNF-α上调,参与银屑病的发病机制[39-40],同样,对前20个关键基因进行转录因子预测后,转录因子RELA调控关键基因的数量最多。因此,RELA可能是参与银屑病病理过程的最重要的转录因子之一,参与到银屑病发病机制中。
综上所述,RELA-STAT1-miRNA-3064-5p很有可能作为新兴调控网络参与银屑病的发生发展,为了证明这个观点仍需要进一步的体外和体内水平的实验验证。
