正庚烷异构化反应研究进展
2023-02-02马爱增李金芝
郭 凯, 马爱增, 李金芝
(中石化石油化工科学研究院有限公司,北京 100083)
烷烃异构化反应是石油炼制工业中重要反应过程之一,在高辛烷值汽油生产、柴油精制降凝、改善润滑油低温流动性等领域得到广泛应用[1-3]。随着国家环保标准的日益提高以及汽车发动机工业的快速发展,车用汽油新标准对汽油池中高辛烷值组分烯烃和芳烃含量进一步限制[4]。异构烷烃作为另一种高辛烷值组分可以有效弥补因汽油池中烯烃和芳烃含量限制而造成的辛烷值损失,是一种理想的汽油调合组分,将直馏汽油和重整抽余油等轻石脑油中正构烷烃异构化的技术近年来得到越来越广泛的关注[5-7]。
目前C5~C6正构烷烃异构化反应的研究已经很成熟,多项技术达到清洁燃料生产要求并已实现工业化,提高了汽油池中前端辛烷值,一定程度上解决了汽油池中异构烷烃不足的问题[8-10]。受生产原料来源限制和高质量汽油需求的驱动,异构化技术有应用于正庚烷及以上长直链烷烃的趋势[11-12]。正庚烷在石脑油中含量较高,其同分异构体的支链化程度越高,辛烷值越高,其中三取代的2,2,3-三甲基丁烷的研究法辛烷值可达112。通过异构化反应实现正庚烷转化为多取代同分异构体可以大幅度提高组分的辛烷值,但当将C5/C6异构化技术应用于正庚烷异构化时,由于裂解副反应的发生导致异构体的选择性严重下降,目标产物收率下降,氢耗增加,以至于目前正庚烷异构化反应的研究还停留在实验室阶段,实际工业应用进展缓慢[13-14]。正庚烷异构化反应仍是国内外学者的研究热点[15-16]。
笔者总结了近年来国内外关于正庚烷异构化反应在催化反应机理和催化材料等方面的研究进展,并提出了该技术的未来发展方向。
1 正庚烷异构化反应机理
自20世纪60年代Kramer等研究正庚烷在AlBr3-H3PO4催化剂上的反应机理开始,学者们对正庚烷异构化反应机理已做了大量研究[17]。通常情况下,正庚烷异构化反应是临氢、高温、连续的气相反应,除生成异构C7烷烃产物外,还产生以丙烷和丁烷为主的低碳烷烃裂化副产物[18-19];其评价在固定床微型反应器中进行,不同催化剂和反应条件下,异构化反应路径有一定差别,目前研究最多的是临氢条件下正庚烷在负载金属-酸载体双功能催化剂上的异构化反应过程。
1.1 经典双功能异构化反应机理
经典双功能异构化机理是指正庚烷在负载金属-酸载体双功能催化剂上发生的异构化反应过程,一般情况下金属中心作为脱氢-加氢中心,而酸中心作为骨架异构或裂化中心[20]。具体反应过程如下:
碳正离子形成:
n-C7H16↔n-C7H14+H2
(1)
(2)
(3)
碳正离子骨架异构化:
(4)
异构烷烃形成:
(5)
i-C7H14+H2↔i-C7H16
(6)
异构化反应是连续发生的,一般认为金属中心上发生的是正构烷烃脱氢生成正构烯烃(见式(1))和异构烯烃加氢生成异构烷烃(见式(6))的反应;而在载体酸中心上发生的是正构烷基碳正离子的形成(见式(2)和(3))、烷基碳正离子骨架异构化(见式(4))和异构烷基碳正离子形成异构烯烃(见式(5))的过程。烯烃由金属中心转移到载体酸中心是通过扩散过程完成的[21]。特别提到的是如采用酸性特别强的超强酸为酸性载体时,最初的碳正离子可能由碳鎓离子分解而来[22]。双功能异构化反应具有如下特点:当金属负载量超过一定量时(如:Pt质量分数大于1.0%),脱氢-加氢步骤在氢气存在的条件下达到平衡,烷基碳正离子在载体酸中心上的骨架异构化过程成为反应的速控步骤,通过调控金属负载量很容易使反应达到脱氢-加氢平衡,因此研究烷基碳正离子的转化在异构化过程中十分重要[23-24]。酸性载体上存在Brönsted酸(B酸)中心和Lewis酸(L酸)中心,通常在金属-酸载体双功能催化剂上,以B酸催化途径为主。近年来围绕着酸性载体上碳正离子中间体的生成与重排等过程,学者们提出了许多观点,归纳总结前人的研究[25-26],正庚烷异构化反应主要有传统单分子异构化反应机理和双分子异构化反应机理2类观点。
1.1.1 传统单分子异构化反应机理
单分子异构化反应机理一般发生在碳链碳数大于4的烷烃异构化反应中,碳正离子的重排主要有经典碳正离子直接重排方式和质子化环丙烷(PCP)中间体重排方式。经典的碳正离子直接重排是指碳正离子通过一系列的氢转移、甲基转移形成其他碳正离子异构体,而不经过环状过渡态过程[27]。质子化环丙烷中间体碳正离子最早由Brouwer等[28]在1972年提出并用于对烷烃异构化反应中碳正离子转化的解释。图1为单分子机理中质子化环丙烷中间体碳正离子异构化过程[29]。单分子异构化反应中包括的异构化过程有2类:A型异构化过程和B型异构化过程。A型异构化过程只改变侧链的位置但不改变烷烃碳链的支链程度,具体包括氢转移和甲基转移过程。B型异构化过程改变烷烃碳链的支化程度,反应是通过形成角位质子化环丙烷正离子中间体,然后再通过角-角质子转移到没有烷基侧链的碳原子上再形成质子化环丙烷环,进而再开环而完成的。A型异构化过程不涉及角位质子的转化,其反应速率大于B型异构化过程。
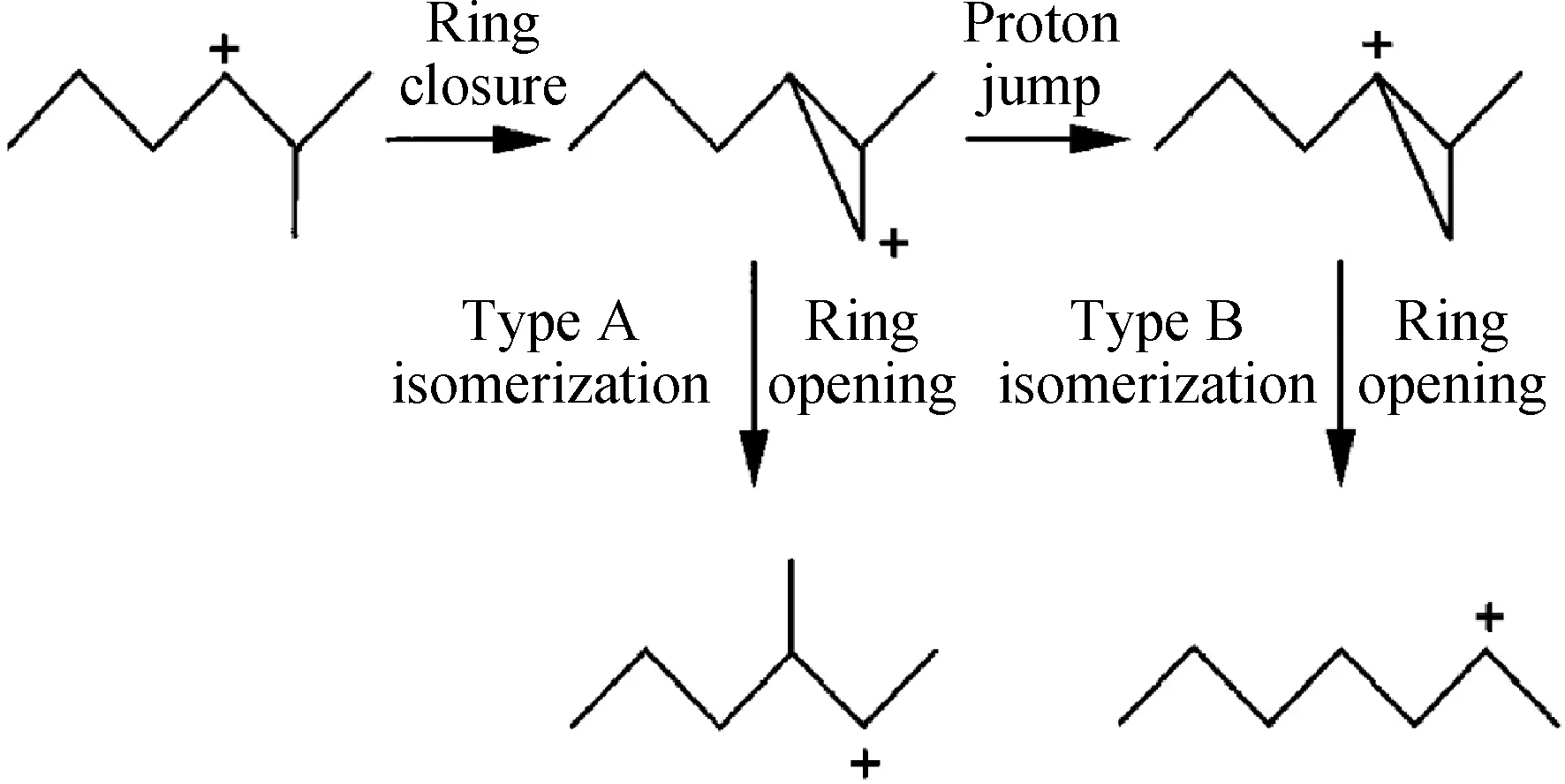
图1 单分子机理中质子化环丙烷中间体碳正离子异构化过程[29]Fig.1 Isomerization process of protonated cyclopropane intermediate carbocation in monomolecular mechanism[29]
Dhar等[30]利用经角位质子化环丙烷碳正离子(CPCP)机理解释了正庚烷通过异构化反应形成单支链和多支链产物的过程,如图2所示。此机理可以对正庚烷在双功能催化剂上的异构化反应进行合理的解释,其中H+向角位的伯碳位的跃迁是反应的速控步骤(c过程和d过程)。CPCP机理存在的问题是过程中角位的H+跃迁目前还没得到证实。通过理论计算表明,质子化得到环丙烷离子并非是异构化反应中的中间产物,而只是一种过渡态[23]。除3-乙基戊烷外,所有正庚烷的同分异构体都可以通过不同碳正离子中间体经CPCP机理而形成[31],过渡态的形成难易导致不同异构体产物分布的不同。实际反应中总会有3-乙基戊烷的生成,因此Mihlyi等[32]和Weitkamp等[33]提出了角位质子化环丁烷机理(CPCB),解释了正庚烷在异构化反应过程中乙基产物的形成。Martens等[34]研究了长链烷烃在Pt/CaY和Pt/USY催化剂上的临氢异构化产物分布,也认为除经CPCP机理反应外,还可以通过大于三元环的碳正离子角位质子化环烷烃机理方式进行反应。
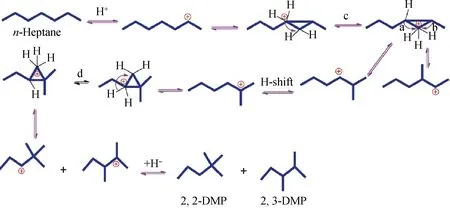
DMP—Dimethylpentane; a, b, c, d—Hydrogen transfer process at different positions图2 正庚烷通过角位质子化环丙烷碳正离子(CPCP)机理异构化过程示意图[30]Fig.2 Schematic diagram of n-heptane isomerization through corner protonated cyclopropane alkylcarbenium ion (CPCP) mechanism[30]
1.1.2 双分子异构化反应机理
双分子异构化反应机理最初是用于解释不能通过PCP途径实现异构化反应的原因,如丁烷的异构化。Agudo等[35]在研究CrHNaY催化剂上正庚烷裂化反应时提出了双分子异构化反应和裂化反应的途径。Blomsma等[29,36-37]在研究不同Pd负载量的Pd/Hβ分子筛催化剂上正庚烷异构化反应和裂化反应机理时提出,当Pd负载量较小时,由一分子正庚基碳正离子与一分子正庚烯通过双分子反应途径形成十四烷基碳正离子,形成的十四烷基碳正离子通过A型异构化过程使烃基骨架发生异构,然后通过A型β-裂解反应生成异庚烷或裂解为小分子产物,双分子反应和单分子反应过程如图3所示。Chao等[38]在研究Pt/MOR和Pt/β催化剂作用于轻石脑油异构化反应时,也采用双分子反应机理来解释正庚烷的裂化产物中含有C3~C6的问题。需要指出的是,单分子反应发生的速率远远快于双分子反应发生的速率,双分子反应机理在长链烷烃异构化中并不占有主要地位,反应过程中并没有检测到双分子中间体的存在。
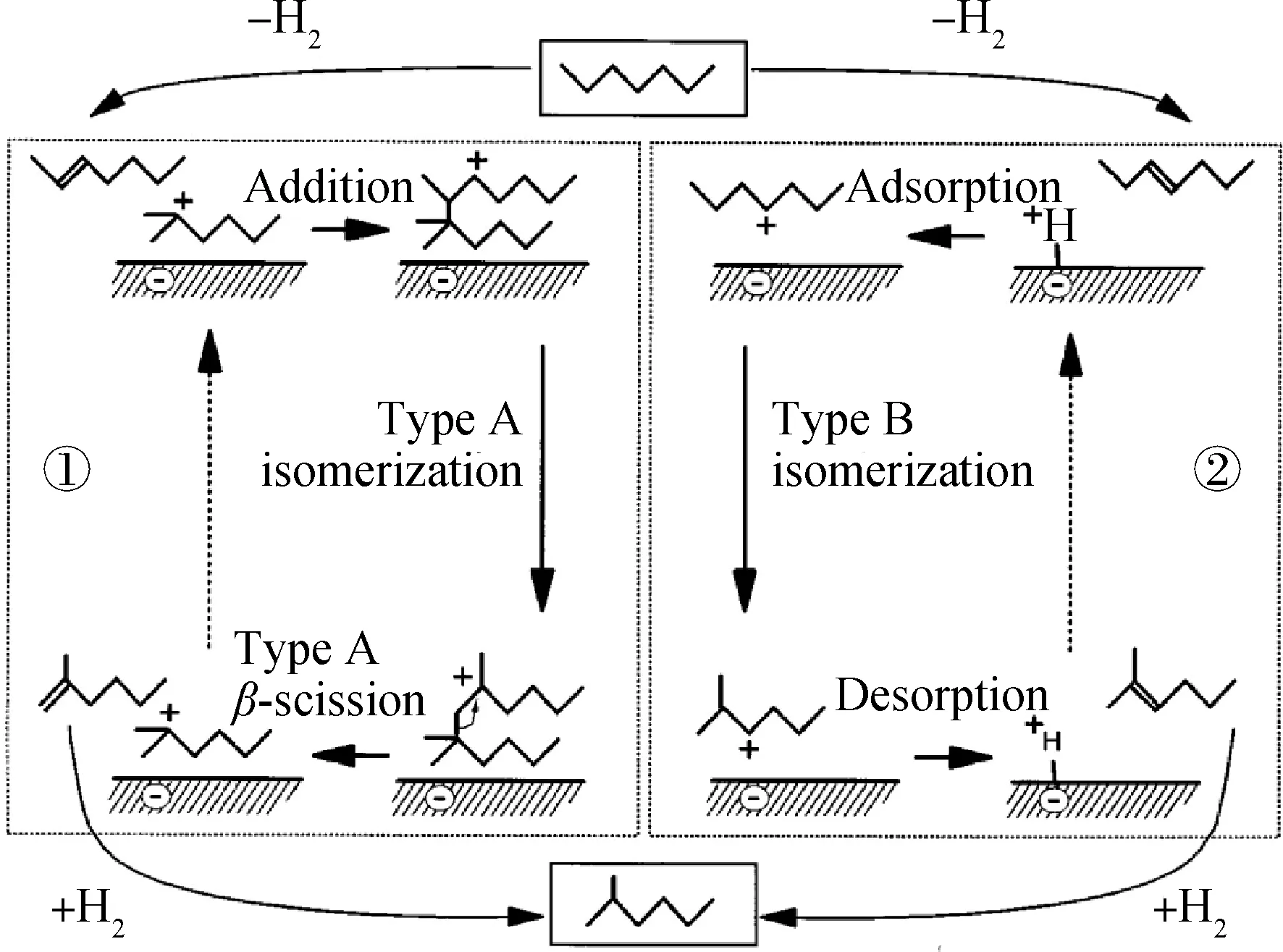
①—Bimolecular isomerization mechanism; ②—Monomolecular isomerization mechanism图3 正庚烷在双功能催化剂上的反应机理[29]Fig.3 Reaction mechanism of n-heptane on bifunctional catalysts [29]
1.1.3 催化剂空间结构效应
分子筛类多孔材料由于其特殊的几何构型及由此种构型所产生的扩散效应,起到控制反应方向的作用,称之为择形性[39]。正庚烷异构化反应中,一维十元环分子筛择形性表现最为明显,归因于这些沸石的孔道直径与庚烷分子的动力学直径相近。适用于正庚烷加氢转化的择形选择性有反应物选择性、产物选择性和过渡态选择性,如图4所示[40]。
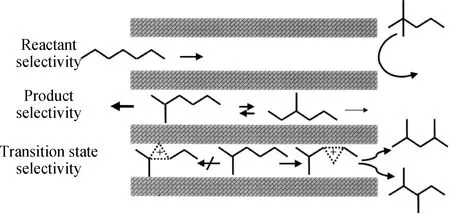
图4 适用于正庚烷加氢转化的择形选择性概念[40]Fig.4 Shape selectivity concepts valid forn-heptane hydroconversion[40]
Poursaeidesfahani等[41]研究了3种不同孔径的分子筛型催化剂(MFI型、MEL型和*BEA型沸石)上产物选择性对正庚烷转化的影响,发现MFI型和MEL型分子筛上双支链异构体的扩散障碍了这些分子向气相的转移,导致了更多裂化反应的发生而使产物分布不同,而*BEA型分子筛不受此影响,产物选择性是*BEA型沸石与MFI型和MEL型沸石的产物分布存在显著差异的主要原因。Sastre等[42]采用ZSM-48和Theta-1型2种一维十元环分子筛用于研究正庚烷及其异构体在孔道内的扩散速率,结果显示ZSM-48由于孔道直径稍大,扩散速率高于Theta-1分子筛,在从单甲基异构体到二甲基异构体的反应中观察到产物选择性的影响。Maesen等[40]研究了正庚烷在TON、MTT、AEL型分子筛上的异构化反应,得出了末端甲基产物的高选择性归因于产物择形选择而邻甲基支链烷烃的低选择性归因于过渡态选择性的结论。
需要指出的是,存在“超笼”或“侧袋”结构的分子筛催化剂对正庚烷异构化选择性不利,原因是其空间效应使多支链异构产物难于扩散离开催化剂孔道而造成裂解副反应的发生,从而导致多支链异构体选择性下降,这是Pt/EU-1催化剂在正庚烷异构化反应中表现出相对较低异构体产率的原因,即其存在1个十二元环的“侧袋”结构[31]。Noh等[43]最近研究了烷烃异构化反应中分子筛结构对中末端甲基异构体选择性的关系,结果显示一维十元环分子筛上孔道的空间结构使异构烷烃产物中间体烯烃的浓度梯度变化是造成甲基异构体高选择性的原因,而不是由于“孔口催化”或其他方式[44]等原因。
1.1.4 副反应的发生
正庚烷在双功能催化剂上发生异构化反应的副反应包括金属中心上正庚烷的氢解反应和酸中心上碳正离子的β-裂解反应。通常氢解反应生成C1、C2小分子烷烃,β-裂解反应生成C3、C4小分子烷烃,大多数副反应以后者为主[19]。通常异构体高选择性发生在异构化反应低转化率时,随着转化率增加,副反应β-裂解反应加剧,异构体的选择性迅速下降。图5为碳正离子通过β-裂解反应分解的5种可能变化途径[29]。由图5可以看出,反应速率由大到小顺序为:A型β-裂解反应,B型β-裂解反应,C型β-裂解反应,D型β-裂解反应,由于A型β-裂解反应要在烷烃碳数大于8时才能发生,而D型β-裂解反应的产物中有不稳定的初级碳正离子形成而不易发生,所以正庚烷主要以B型和C型β-裂解反应途径发生反应。在双功能催化剂上,正庚烷按照B型β-裂解反应的速率和按照PCP途径异构化反应的速率相近而大于C型β-裂解反应的速率[45],所以反应过程中一般表现为异构化反应与裂化反应的竞争关系。

图5 碳正离子通过β-裂解反应分解的5种可能变化[29]Fig.5 Five possible variants of carbonium ion decomposition through β-scission reaction[29]
1.1.5 异构化反应与副反应同时存在时的反应机理
正庚烷异构化反应进行时,裂解反应总是伴随着异构化反应同时发生,近年来许多学者对2种反应路径进行了研究并提出了以下2种主要观点。
第一种观点认为:正庚烷首先异构成单甲基支链异构体,再异构化为多支链异构体,最后多支链异构体发生裂解副反应生成小分子副产物。Patrigeon等[46]评价了不同分子筛催化剂正庚烷异构化反应性能并研究了单甲基支链异构体、多支链异构体和裂解小分子的产率与正庚烷转化率的关系,发现在Pt/Hβ催化剂上正庚烷是按照先转化为单甲基支链异构体再转化为多甲基支链异构体的路径进行反应的。
第二种观点认为:正庚烷首先异构化生成单甲基支链异构体,单甲基支链异构体异构化为多支链异构体的同时,所有支链异构体同时会发生裂解反应生成裂解小分子[47]。图6为第二种观点进行异构化反应时的路径。根据β-裂解反应机理,单甲基取代庚基碳正离子通过β-裂解反应后要生成更稳定的碳正离子中间体,所以在临氢条件下裂解产物只有正丁烷和丙烷而没有异丁烷(见图5的C型β-裂解反应)。单甲基取代的庚基碳正离子通过异构重排生成二甲基取代庚基碳正离子,二甲基取代庚基碳正离子发生β-裂解反应后的产物只有异丁烷和丙烷而没有正丁烷(见图5的B型β-裂解反应),所以可以根据裂解产物的生成情况判断反应发生的途径来解释异构化反应机理。WANG等[18]研究了Pt/H3PW12O40/Zr-MCM-41催化剂上正庚烷异构化反应的机理,发现裂解产物中异丁烷产率要高于正丁烷产率,且异丁烷出现在较宽的反应温度范围内,而正丁烷只在较高的温度下形成,证实了较低温度时裂解反应路径是按照第一种观点进行的,而较高温度时是按照第二种观点进行的。正庚烷在大多数酸性载体上的反应路径都是按第二种观点进行的[48]。要使正庚烷异构化选择性提高,必须控制裂解反应的发生[49],需合理调配酸性中心和金属中心的平衡。

图6 双功能催化剂催化正构烷烃异构化机理[50]Fig.6 Mechanism of n-alkane isomerization catalyzed by a bifunctional catalyst [50]
1.1.6 金属中心上氢的转移
Hattori[51]提出了“氢分子-质子酸位点”的概念用来解释Pt负载的固体超强酸催化剂上正庚烷异构化反应中金属与氢的作用,如图7所示。首先H2在金属Pt表面被解离吸附为2个氢原子,氢原子经过溢流到达酸载体表面的酸位上,将电子给予L酸并形成H+,在临近L酸位的氧原子上作为质子酸位,同时另一个氢原子与被L酸捕获的电子形成H-,这样H2分解为H+和H-分别参与烯烃的质子化和烷烃碳正离子的氢转移过程。Nie等[52]采用In2O3掺杂的Pt/WO3/ZrO2催化剂用于正庚烷异构化反应,利用上述机理合理解释了反应过程,通过In2O3的掺杂,提高了催化剂载体上的B酸量,加快了异构碳正离子和氢离子的转移速率,提高了正庚烷的转化率和选择性。
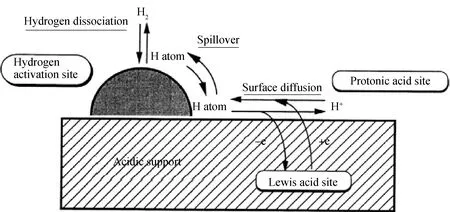
图7 固体超强酸上的氢分子-质子酸位点形成示意图[51]Fig.7 Schematic diagram for the formation of hydrogen molecule-proton acid sites on solid superacid [51]
1.2 金属-环丁烷反应机理
郭燮贤等[53]最初研究金属催化剂上季碳异构产物的键转移和重排过程时,根据反应产物分布规律归纳总结并提出了烷烃异构化的类三元环机理,即金属-环丁烷反应机理。包括3次甲基吸附态的生成和环转移过程2个步骤。Blekkan等[54]采用金属-环丁烷反应机理解释了氧修饰的碳化钼催化剂上正庚烷临氢异构化反应过程,如图8所示。反应物先与金属位形成环状过渡态,然后通过碳-碳键断裂生成一分子烯烃和一分子金属-烯化合物,通过不同位置碳-碳键重新结合完成骨架异构,生成相应的异构产物。理论上该反应得到2-甲基己烷与3-甲基己烷的比例接近相等,实际反应中3-甲基己烷的收率高于2-甲基己烷,这可能是其中间过渡态更容易形成造成的[55-56]。金属-环丁烷反应机理虽然研究的较早,但近年来的研究逐渐减少,其中的一些过程如裂化产物的产生等还没有明确的描述。

×1—Single forming position; ×2—Double forming positions; M—metal site图8 正庚烷异构化过程中金属-环丁烷中间体的键转移机理示意图[54]Fig.8 Schematic diagram for the bond transfer mechanism of metal-cyclobutane intermediate during n-heptane isomerization[54]
2 正庚烷异构化反应催化剂
20世纪60年代,从Hepp等[14]将Friedel-Crafts催化剂(主要为AlCl3/HCl)用于正庚烷异构化反应开始到现在,许多学者一直致力于正庚烷异构化反应催化剂的研发。到目前为止,用于正庚烷异构化反应研究的催化剂包括Friedel-Crafts催化剂、金属化合物类催化剂和金属-酸载体双功能催化剂3类。
2.1 Friedel-Crafts催化剂
Friedel-Crafts催化剂主要由卤化铝和助催化剂卤化氢等组成,其用于正庚烷异构化时虽具有较高的活性,但同时会发生明显的副反应而使异构产物选择性急剧下降,因此其目前用于正庚烷异构化反应的研究已经很少。
2.2 金属化合物类催化剂
用于正庚烷异构化研究的金属化合物类催化剂主要包括钼、钨的碳化物和部分还原氧化物。此类催化剂往往表现出高选择性,但其较低的活性和较高的反应温度对产物中多支链异构体的生成不利[57]。
Blekkan等[54]在流动的空气气氛条件下使Mo2C变成部分氧化的碳化物MoOxCy并用于正庚烷异构化反应,发现其具有较好的活性和选择性,其中异构化产物以2-甲基己烷和3-甲基己烷为主,并据此提出了如图8所示的金属-环丁烷反应机理。李学斌等[58]制备了β分子筛负载碳化钼催化剂,以正庚烷为模型反应物采用连续流动固定床反应装置考察了温度、压力、空速和氢/烃比对β-Mo2C/β分子筛临氢异构化性能的影响,得到正庚烷转化率为82%、异构化产物选择性和收率分别达到71%和58%的反应结果。Ribeiro等[59]发现经氧化处理的WC和β-W2C对正己烷、正庚烷异构化反应具有高催化活性,几乎没有发现环化和氢解反应的痕迹。
2.3 金属-酸载体双功能催化剂
金属-酸载体双功能催化剂是一类主要由活性金属组分和酸性载体组分组成的催化剂,一般是在氢气存在的条件下使用,金属组分提供金属中心,酸载体提供酸性中心,金属中心和酸性中心的匹配是此类催化剂具有高活性、高选择性的关键[60]。
2.3.1 活性金属组分
双功能催化剂上金属组分主要由第Ⅷ族元素的Pt[60-61]、Pd[62-64]和Ni[65-67]等金属或其化合物的一种或几种组成,第ⅡB族和稀土元素常常作为助催化剂而被加入[68]。金属一般以团簇的形式负载分散在酸性载体上[69],常用的负载方法为浸渍法和离子交换法[70]。金属的负载量、粒径、分散度等对正庚烷异构化反应有直接影响。
2.3.2 酸载体
目前用于正庚烷异构化反应研究的载体主要有氧化物类和固体酸类载体。固体酸类载体包括分子筛、固体超强酸和杂多酸等材料,其具有酸性强、产物易分离、腐蚀小等优点,是广泛研究的一类材料。
(1)氧化物类载体
氧化铝(Al2O3)和氧化硅(SiO2)是常用的氧化物载体,通常作为异构化催化剂载体,需要进行处理以提高其酸性,如采用AlCl3处理Pt/Al2O3得到Pt/Al2O3/Cl催化剂,成功实现了C5~C6烷烃异构化工业化[71]。近年来也有报道金属负载酸性氧化物用于正庚烷异构化反应研究。Samad等[72]将Pt负载到酸性Al2O3-SiO2载体上并调整双功能催化剂上金属中心与酸中心的距离成功制备了不同的Pt/Al2O3-SiO2催化剂并用于正庚烷临氢异构化反应研究。结果发现,金属中心与酸中心距离为纳米尺度的双功能催化剂在正庚烷异构化反应评价中具有更好的催化性能。氧化物类载体通常需要较高的反应温度,但正庚烷异构化反应是微放热反应,高温对异庚烷的生成不利,限制了其应用。
(2)分子筛类载体
以分子筛为酸载体的双功能催化剂是性能优异的一类异构化催化剂。其具有在宽范围内可调变的酸性和化学组成、高热稳定性、分子筛选性质,高原料杂质耐受程度以及可完全再生等优点。微孔人工合成沸石是正庚烷异构化反应常用的载体,其孔径一般小于2 nm,落在分子尺度范围内,有筛选分子的特性。表1为常用于正庚烷异构化反应的分子筛载体的基本结构。归纳起来有十二元环分子筛(大孔)与十元环分子筛(中孔)两类。十二元环分子筛主要包括Y、β、SAPO-5和MOR型分子筛,这类分子筛的孔径大于庚烷分子在反应条件下的动力学直径,其结构对庚烷不具有择形作用。十元环分子筛主要包括ZSM-5、ZSM-22、EU-1、ZSM-23、ZSM-48、SAPO-11、SAPO-31和SAPO-41等。此类分子筛的孔径与反应条件下庚烷分子的动力学直径相当,其结构对庚烷具有择形作用。
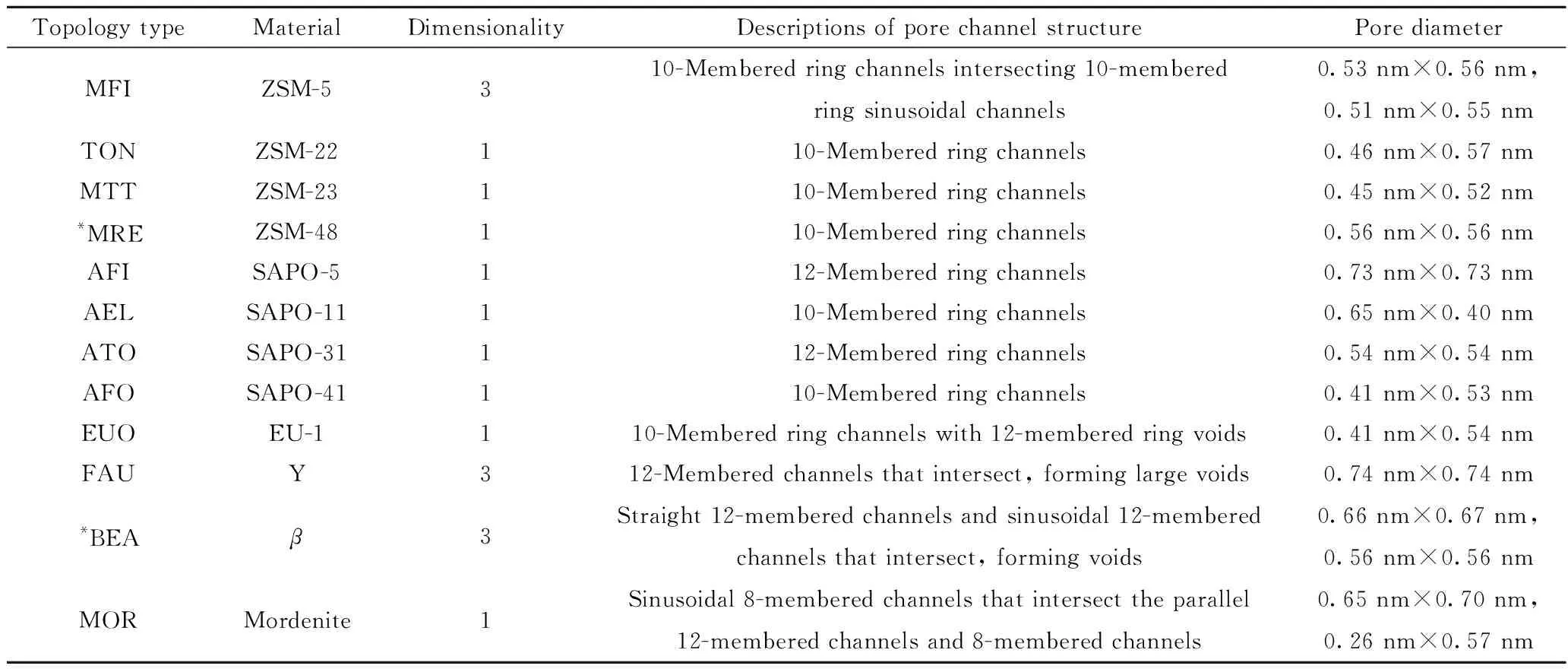
表1 常用于正庚烷异构化反应的分子筛类载体基本结构Table 1 Basic structure of zeolite supports commonly used in n-heptane isomerization
以十二元环分子筛为载体的催化剂在异构化反应中整体表现出较高的活性,反应温度一般不超过300 ℃,其中,Y型和β型分子筛具有三维孔道和“笼”结构,反应温度比MOR和SAPO-5型分子筛要低。通常由于上述载体的孔径较大,多支链的庚烷异构体在孔道中不受扩散限制,在高转化率时多支链庚烷异构体具有高收率,但多支链中间体的增加又会导致高裂化率,庚烷异构体收率与裂化小分子产物收率矛盾较大。若要获得高多支链异构体收率,酸中心和金属中心应尽可能彼此接近,以避免烯烃转移速率对反应速率的限制,同时金属中心应具有较高的加氢-脱氢活性,以使酸中心具有足够数量的烯烃,并使不饱和产物快速氢化为烷烃[25]。
三维孔道结构的十元环分子筛,如ZSM-5等,由于孔口直径的限制,多支链庚烷异构体很难扩散到其孔道内的酸中心上,而正庚烷的扩散不受限制。当正庚烷扩散到其直筒型和正弦型孔道交叉处的孔隙中时足以使正庚烷连续反应转化为多支链异构体,而其相对窄的孔道限制了多支链异构体的扩散,导致严重的裂解反应,使多支链异构体进一步生成小分子产物而离开分子筛孔道,异庚烷选择性大幅度降低,此类分子筛不适合作为正庚烷异构化反应的载体[70,73]。一维孔道结构的十元环分子筛,如ZSM-22、ZSM-23、和部分SAPO系列等不具有交叉孔隙结构,相比于三维孔道结构十元环分子筛载体,形成多支链中间体的能力弱,但往往表现为低裂化率。一般情况下此类载体在高转化率下单支链异构体收率很高,但多支链异构体收率极低[64],此外,此类载体往往酸性弱于十二元环分子筛,需要较高的反应温度[74]。
近年来,报道了许多以分子筛和介孔材料结合而成的复合材料为载体用于正庚烷异构化反应的研究。常用的介孔材料以硅基介孔材料为主,如MCM系列[75-76]、SBA系列[77]等,这类材料通常具有较大的比表面积和孔径、规整的孔道结构,有利于较大分子的活化及扩散,但其在高压下的水热稳定性差、酸密度小,其本身不利于在正庚烷异构化反应中的应用。通常采用具有酸性的分子筛复合介孔材料或在其骨架中引入金属离子而产生酸性中心[78],强化了分子筛的扩散能力和介孔材料的酸性,表现出优异的正庚烷异构化性能。表2为以分子筛及其复合材料为载体的双功能催化剂在正庚烷异构化反应中的反应条件与性能比较。
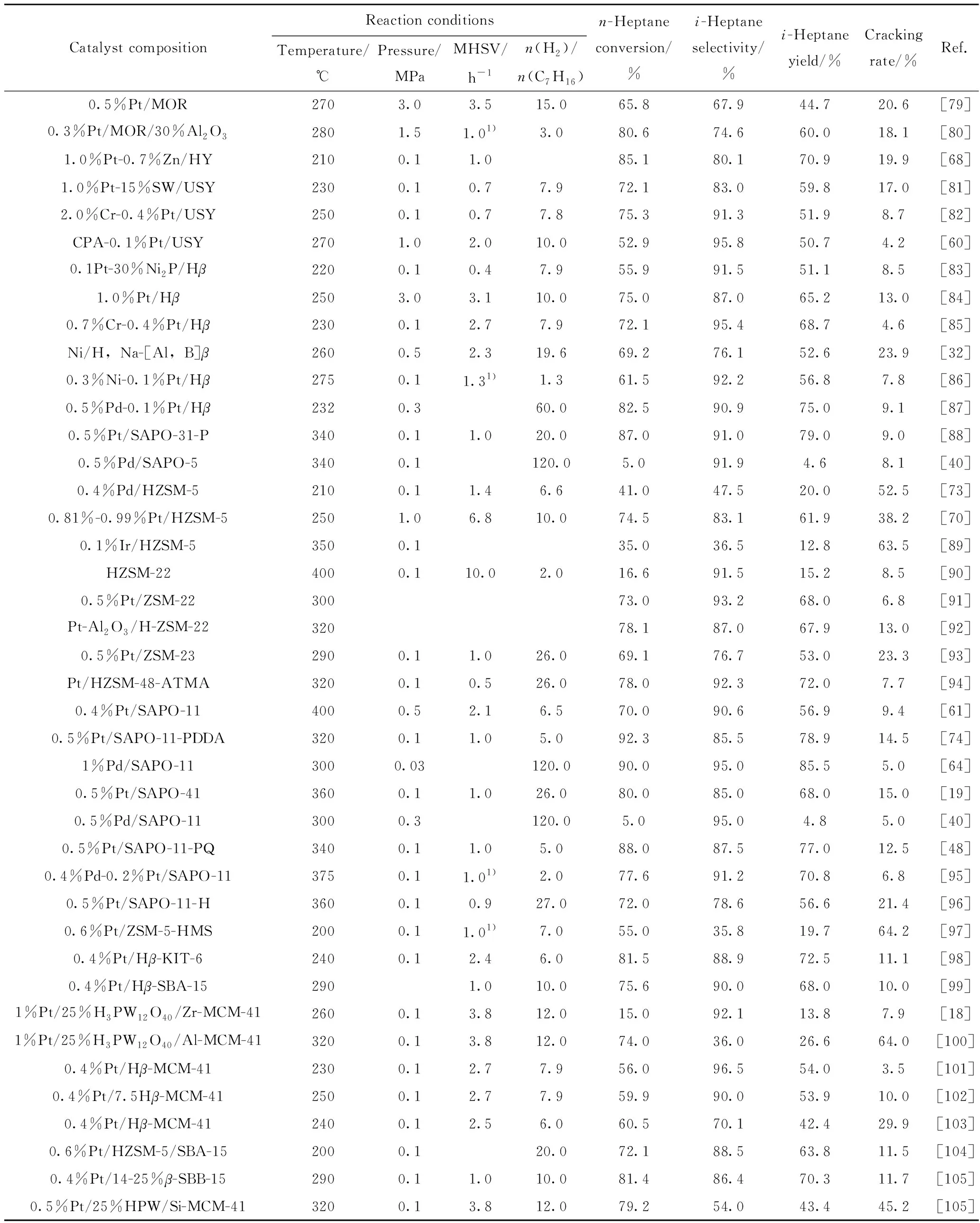
表2 以分子筛及其复合材料为载体的双功能催化剂在正庚烷异构化反应中反应条件及性能比较Table 2 Comparison of reaction conditions and properties of bifunctional catalysts with zeolite and its composites as supports in n-heptane isomerization
(3)固体超强酸类载体
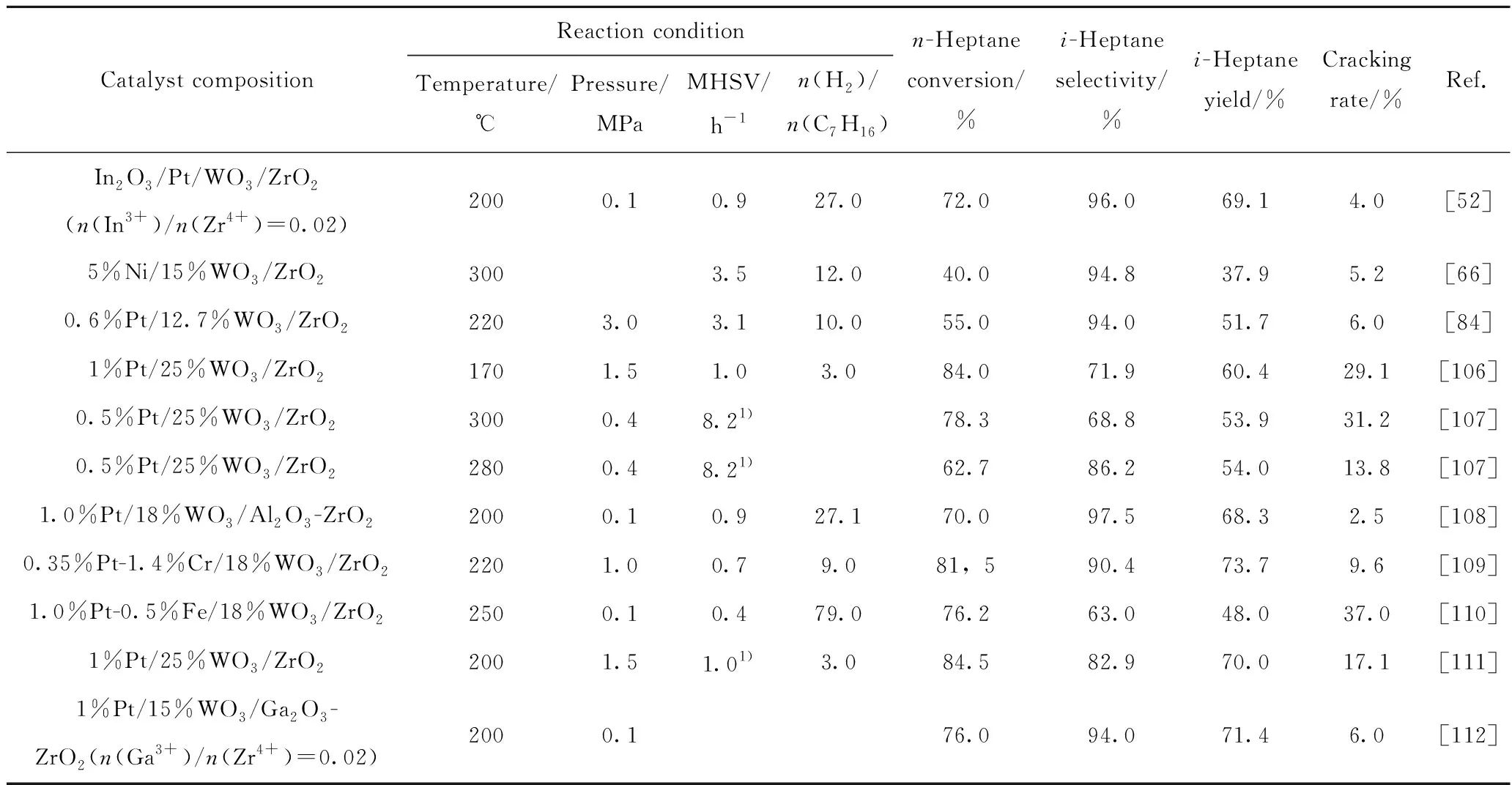
表3 以固体超强酸为载体的双功能催化剂在正庚烷异构化反应中的反应条件及性能比较Table 3 Comparison of reaction conditions and properties of bifunctional catalysts with solid superacids as supports in n-heptane isomerization
Shkurenok等[106]评价了1.0%Pt-(10%~35%)WO3/ZrO2催化剂催化正庚烷异构化反应性能,结果显示,在170 ℃下,质量分数25%WO3条件下,正庚烷转化率达到84%,异构体选择性达到71.9%。Jermy等[107]在不同条件下制备了Pt/WO3-ZrO2催化剂,结果发现,除已知WO3负载量、煅烧温度外,包括水合ZrO2的预干燥温度、浸渍方式、偏钨酸铵浸渍液的pH值、煅烧气氛、铂源等都在一定程度上影响正庚烷异构化反应的活性和选择性。
(4)杂多酸类载体
用于正庚烷异构化研究的杂多酸类载体主要是具有Keggin结构的磷钨酸(H3PW12O40)和硅钨酸(H4SiW12O40)。杂多酸的酸性较强但比表面积较小,一般需要负载在其他载体上。负载金属的杂多酸双功能催化剂在烷烃异构化活性和选择性方面具有优异的表现。
汪颖军等[113]制备了介孔沸石负载杂多酸催化剂Ni-H4SiW12O40/MCM-41并用于正庚烷异构化反应,结果发现4%Ni/30%H4SiW12O40催化剂在反应温度300 ℃、反应时间6 h的条件下性能最好,正庚烷转化率为18.5%,异构化选择性为74%。Fuente等[114]合成了Pt/H3PW12O40/SBA-15催化剂并考察了其正庚烷临氢异构化反应性能,结果发现通过还原处理后,杂多酸分散更好、Pt纳米粒子更小;其最佳组分为1%Pt/20%H3PW12O40,在反应温度为320 ℃时,正庚烷转化率为56.7%,异构化选择性为100%,高异构化选择性的主要原因归因于Pt纳米晶体化、适当的酸强度与酸平衡及高度有序的介孔。
3 结语与展望
调研了正庚烷异构化反应的反应机理和催化材料,反应机理包括经典的双功能异构化反应机理和金属-环丁烷反应机理;催化材料包括Friedel-Crafts型、金属化合物型和金属-酸性载体双功能催化剂。
(1)经典的双功能异构化反应机理是目前研究广泛的反应机理,对正庚烷的催化化学反应过程具有详细的解释,形成了成熟的理论,对解决当前正庚烷异构化催化剂活性与选择性之间的矛盾和研究开发新型催化剂具有重要理论指导意义。
(2)金属-酸载体双功能催化剂是应用最广泛的催化剂,其具有选材广泛、金属-酸平衡调整方便、反应条件温和等优势。载体的种类不同,正庚烷的转化率和异构体的选择性不同。十二元环分子筛载体上,多支链异构体的选择性较高,但正庚烷转化率较低、裂化率较高。固体超强酸类载体在合适的条件下,可以在较高的转化率下仍具有高异构体选择性,但条件苛刻,稳定性稍差。
(3)虽然对正庚烷异构化反应机理已有大量研究报道,但对于催化反应路径、反应原理、烷烃分子活化转移、碳链断裂与重排等仍需不断研究与探索。目前对于金属-酸载体双功能催化剂研究最为广泛,其中以分子筛为载体的催化剂,表现出很好的异构化前景。当前催化剂的研发关键是满足正庚烷在高转化率下仍具有高异构体选择性。通过调整金属组分种类、含量及分散度,扩孔或采用微孔与介孔材料制成复合材料等调控手段,合理调控载体酸性功能和金属中心金属功能的协同作用是正庚烷异构化反应研究的方向。
