非晶合金Fe3Ni3的轨道杂化及成键性质研究
2022-12-27郑新喜方志刚曾鑫渔侯欠欠刘立娥
郑新喜,方志刚,曾鑫渔,侯欠欠,刘立娥
(辽宁科技大学 化学工程学院,辽宁 鞍山 114051)
随着能源危机的加剧,化石能源逐渐无法满足当今时代发展需求,同时由于碳排放造成的温室效应已经成为各国不可忽视的环境问题,因此迫切需要寻找一种新型能源来解决发展中的环境保护问题。在化石能源危机出现后,氢能以其热值高、无污染、利用形式多和安全性好等特点成为未来环保能源发展的热点。目前氢能主要来源于电解水制氢,但传统的铂碳催化剂成本高昂、催化效率较低,应用效果并不理想,因此需要开发新型催化剂。
与传统的晶态合金相比,非晶合金以其短程有序、长程无序的特点在催化[1-2]、储氢[3-4]和磁学性质[5-6]等方面展现出更优异的性能,自20世纪中叶被发现以来,便受到新型材料科学领域研究人员的高度重视,该新型材料的各种综合性质也得到了广泛研究。其中过渡金属原子由于其本身原子d轨道电子未满的特殊性,能够在原子轨道杂化方面展现出与主族金属元素不同的杂化方式,吸引了广大科研人员的注意。在非贵金属原子中,Fe基[7-8]与Ni基[9-11]组成的非晶合金,具有价格低廉、安全无毒和物理化学性能优越等特点,符合绿色化学发展趋势,成为目前非晶合金研究的热点内容,例如针对Fe基构筑的Fe-Cr[12]、Fe-Co[13-14]体系及以Fe基、Ni基构成的Fe-Cr-Ni[15-16]体系等进行的研究,均已取得显著突破。其中WANG等[17]发现基于N掺杂石墨化碳层(NCL)的Fe-Ni系催化剂在电解水阳极析氧(OER)反应中的催化性能与稳定性均超过基准RuO2催化剂;郑冰霞[18]研究发现,二元体系非晶态Fe-Ni合金在金属-有机框架材料(MOF)中的电解水OER性能表现突出;MA等[19]研究发现,OER反应中非晶态Fe-Ni-Co三金属杂化催化剂在电流密度为10 mA/cm2下表现出过低电位(245 mV),其稳定性更是达到 12 h以上;CHEN等[20]发现Fe-Ni合金可经熔融纺丝法制备成表面质量良好的完全非晶结构合金,这为Fe-Ni非晶合金的制备提供了新的研究方向。
宏观催化过程中的催化性能及稳定性主要通过宏观实验进行测定,该体系催化剂的微观局域结构模型、内部原子间成键及轨道杂化等基本性质亟待解决,其微观原子轨道理论及成键性质的研究目前鲜有报道,因此难以对其宏观性质进行理论解释。YANG等[21]研究发现,当电催化剂内部Fe和Ni 原子比为1:1时,对OER反应更为有利。
本文根据前人的研究成果设计团簇Fe3Ni3局域结构模型,并计算出该团簇稳定空间构型,从态密度与成键两方面对团簇Fe3Ni3展开研究,进一步了解内部轨道杂化方式及成键情况,对现行该体系催化剂在宏观催化过程中表现出的良好稳定性作出一定的机理解释,进而为生产稳定性高、催化能力强的非晶合金催化材料提供理论支撑。
1 模型与理论方法
依据拓扑学原理[22],设计出团簇Fe3Ni3的6个原子所有可能的排列分布典型空间构型,例如常见的平面构型、三棱锥型和四角双锥型等空间构型。根据团簇自旋多重度理论,在单、三重态下进行优化计算,以找到稳定的优化构型。
在过渡金属体系的密度泛函计算中,本文选择B3LYP泛函,其是众多科研工作者采用的默认泛函,运算时综合表现较为优异。由于本文研究的Fe-Ni体系的两种原子均为第四周期VⅠⅠⅠ族过渡金属元素,Lan12dz基组针对其内层电子用Los Alamos ECP赝势,外层电子用DZ基组,计算精度完全达到要求[23]。
综上,本文依据密度泛函理论[24-25](Density functional theory,DFT),在B3LYP/Lan12dz量 子化学水平下,将团簇Fe3Ni3的典型初始空间构型采用Gaussian09软件和Multiwfn软件[26]进行全频率验证和几何构型优化以及数据处理。其中Fe、Ni采用WADT等[27]的含相对论校正的有效核电势价电子从头计算基组,即 18-eECP的双ξ基组(3s、3p、3d/2s、2p和 2d)。
对优化后的结果排除含虚频的不稳定构型和空间结构相近的高能量构型后,最终得到9种稳定的优化构型。优化时,需要满足的优化收敛条件为:最大作用力< 0.00045,均方根作用力< 0.00030,最大位移< 0.00180,均方根位移< 0.00120。以上全部计算采用Gaussian09软件与计算机HP-Z440完成。
2 结果和讨论
2.1 团簇Fe3Ni3稳定构型
得到的9 种优化构型包括5 种三重态构型与4 种单重态构型。将9 种优化构型中具有最低校正能的构型 1(3)作为基准(0.000 kJ/mol),计算其它8种构型的相对能量。在图1中,构型下方括号内数据为该构型的相对能量,序号上标为该构型的自旋重态。以第一个构型为例:1(3)(0.000 kJ/mol)表示该构型相对能量为0.000 kJ/mol,自旋重态为三重态。由图1可知,热力学稳定性由高到低排序为1(3)> 2(3)> 3(3)> 4(3)> 5(3)> 1(1)> 2(1)> 3(1)> 4(1)。三重态优化构型的相对能量均小于单重态,说明三重态构型比单重态构型稳定性更好,其中构型1(3)的稳定性最好,而构型4(1)的稳定性最差,这为该团簇催化剂在现实生产过程中的基础构型选择提供了理论指导。
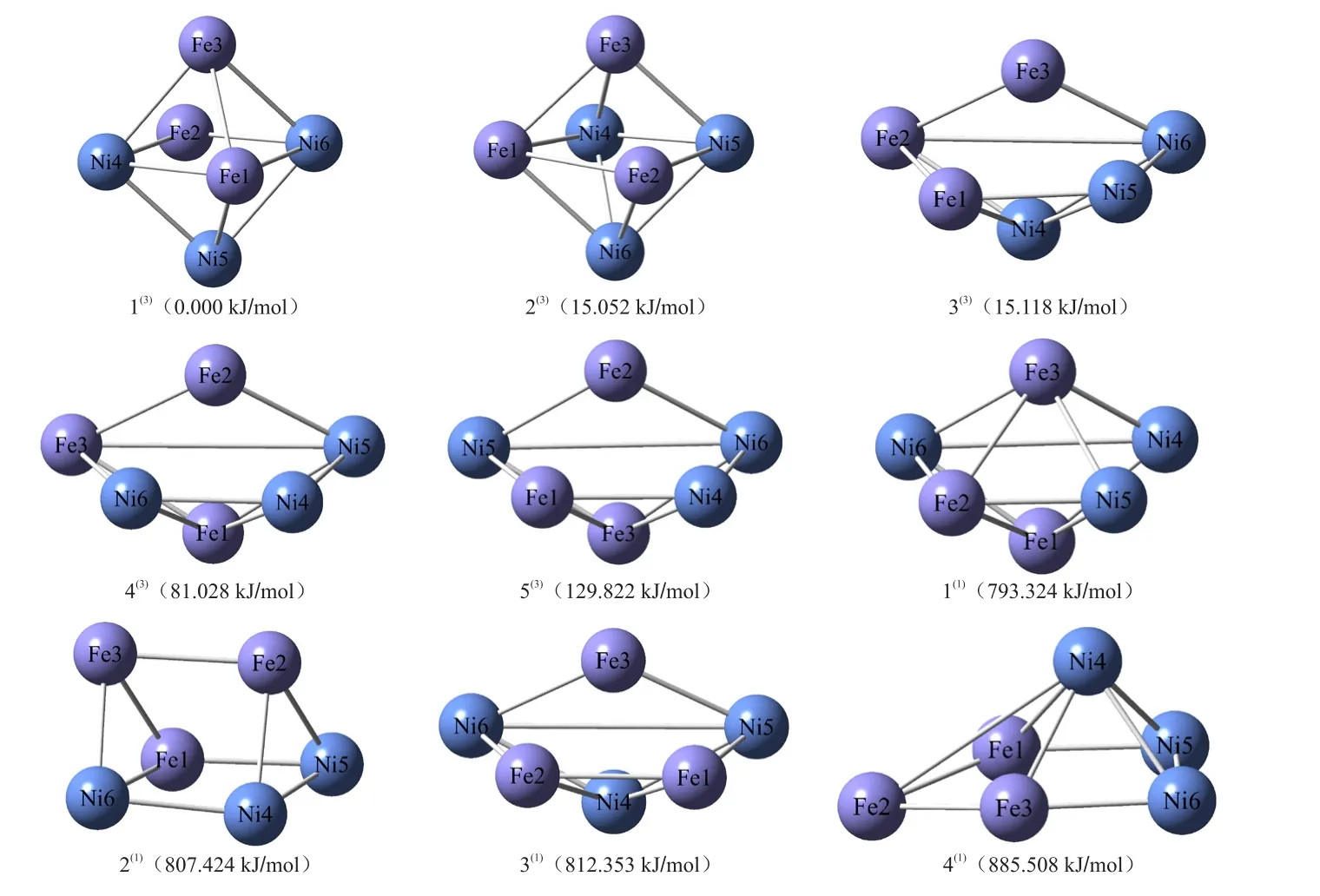
图1 团簇Fe3Ni3的优化构型Fig.1 Optimized configurations of cluster Fe3Ni3
2.2 团簇Fe3Ni3轨道杂化分析
态密度(DOS)指单位能量范围内电子云的分布情况,总态密度(TDOS)指各构型的总体电子分布情况,其中分波态密度(PDOS)表征某一段或某一轨道的电子状态。能带结构的态密度分析具有可视化特征,其分析结论与能带实验结果可以相互印证,且对态密度进行分析相较于能带分析的结果更具有直观性。因此,对团簇Fe3Ni3的各构型总态密度和分波态密度进行分析可以得到该团簇的轨道杂化具体情况,如图2所示。
图2中的竖直虚线表示费米能级(EFermi=费米能级是描述团簇Fe3Ni3在催化反应过程中催化活性的参考能级。费米能级附近的态密度峰值越大,说明团簇在该能量处的轨道杂化作用越强。由图2可知,总态密度中三重态构型的最高峰均在费米能级右侧,而单重态构型的最高峰均在费米能级左侧,说明自旋多重度对团簇Fe3Ni3的轨道杂化具有一定的选择性。
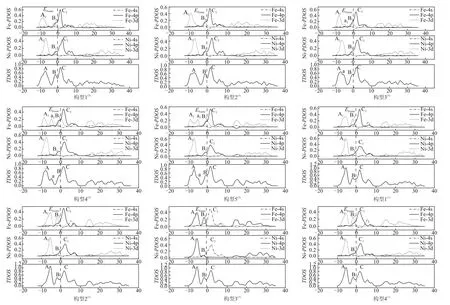
图2 团簇Fe3Ni3优化构型的PDOS和TDOSFig.2 PDOS and TDOS of optimized configurations of cluster Fe3Ni3
对总态密度图中费米能级左右两侧的峰值进行编号(A、B和C),接着按照2.1节结果将三重态构型分为两组,第一组为稳定性较好的构型(构型1(3)与构型2(3)),第二组为稳定性较差的构型(构型3(3)、构型4(3)与构型5(3))。第一组的A峰主要由该构型中Fe-3d与Ni-3d轨道贡献,B峰由Fe-4p、Fe-3d、Ni-4p和Ni-3d轨道贡献,而C峰由Fe-4p与Ni-4p轨道贡献,说明该类构型内部存在3d-3d、3d-4p与4p-4p 3种轨道杂化方式。第二组构型的A峰与第一组相同,由Fe-3d与Ni-3d轨道贡献,但B峰相对于第一组构型与C峰重叠程度增加,基本消失,且在A、B峰之间出现新峰a,均由Fe-3d轨道贡献,C峰由Fe-4p与Ni-4p轨道贡献,说明第二组构型内部主要存在3d-3d与4p-4p两种轨道杂化方式,同时a峰会降低构型稳定性,说明三重态构型内部的4p-3d轨道杂化对构型的稳定性有一定的促进作用。
而对于单重态构型来说,除构型3(1)外,其余3 种单重态构型的A峰均由Fe-3d与Ni-3d轨道贡献,B峰几乎均由Fe-3d轨道贡献,此时Ni-3d轨道贡献基本消失,C峰的主要贡献者是Fe-4p与Ni-4p轨道;而构型3(1)的A峰由Fe-4p与Ni-4p轨道贡献,B峰由Fe-4p轨道贡献,C峰主要由Fe-4s与Ni-4s轨道贡献。针对构型3(1)的特殊杂化方式,推测是在单重态下,其构型空间结构的特殊性使得原子轨道杂化出现改变,对此将进行更加深入的研究。
综上,团簇Fe3Ni3内部主要存在 4s-4s、4p-4p、4p-3d与 3d-3d 4 种轨道杂化方式,其中 4s-4s轨道杂化作用最弱,4p-4p与4p-3d两种轨道杂化作用较强,3d-3d轨道杂化的作用最强,对构型稳定性贡献最大,这也与Fe、Ni两种原子最活泼成键轨道均在d轨道的结论相互印证。
2.3 团簇Fe3Ni3成键分析
团簇Fe3Ni3优化构型中各原子间成键的平均键长如表1所示,键长数值可体现成键与否,若键长小于两原子半径和,表明两原子间成键。原子间键长越短,原子轨道重叠越大,成键越牢固,因此原子间键长是判断原子间成键强度的重要依据之一。依据各不同构型的成键键长数据的标准差(σ,用于分析键长数据分布,标准差越大,数据波动范围越大)和化学键理论[28]得出,Fe—Fe键的键长整体波动范围最小(σ= 0.018),且三重态键长均大于单重态键长,说明Fe—Fe键在单重态下成键普遍强于三重态。Fe—Ni键的键长整体波动范围较大(σ= 0.021),而Ni—Ni键的键长整体波动范围最大(σ= 0.039),说明自旋多重度对构型内部Fe—Ni键、Ni—Ni键具有一定的影响,对Fe—Fe键影响不大。
由表1可知,三重态下,构型 1(3)、2(3)和构型3(3)、4(3)和 5(3)的Fe—Fe键的键长均呈下降趋势,构型2(3)、3(3)呈上升趋势;Fe—Ni键在构型 1(3)、2(3)和 3(3)呈上升趋势,构型3(3)、4(3)和5(3)呈下降趋势。整体来看,三重态下Fe—Fe键与Fe—Ni键变化趋势基本相同,说明二者的相互作用对三重态构型稳定性方面有促进作用。单重态下,Fe—Fe键和Ni—Ni键变化趋势相同,而Fe—Ni键与Fe—Fe键、Ni—Ni键的变化趋势完全相反,说明Fe—Fe键和Ni—Ni键之间存在协同作用,可促进单重态构型的稳定性,而Fe—Ni键与Fe—Fe键、Ni—Ni键之间存在拮抗作用,在一定程度上削弱了单重态构型稳定性。

表1 团簇Fe3Ni3优化构型的平均键长Table 1 Average bond lengths of optimized configurations of cluster Fe3Ni3
团簇Fe3Ni3优化构型中各原子间的平均键级如表2所示,键级指分子中相邻原子成键强度,键级越高,成键强度越大,构型稳定性越强。由表2可知,各构型的Fe—Ni键在单重态、三重态中均为正值,对构型稳定性起促进作用。三重态中除构型3(3)外,其余构型的Fe—Fe和Ni—Ni键平均键级均为正值,说明其余构型各原子间平均键级均对成键有促进作用,而构型 3(3)的Fe—Fe键和Ni—Ni键平均键级对其成键有削弱作用,不利于构型稳定性。单重态中Fe—Fe键平均键级除构型 1(1)外,Ni—Ni键平均键级除构型1(1)、3(1)外,其余均为正值,说明其余构型各原子间平均键级均对成键有促进作用,而构型 1(1)的Fe—Fe键和Ni—Ni键、构型 3(1)的Ni—Ni键的平均键级对其成键均有削弱作用。
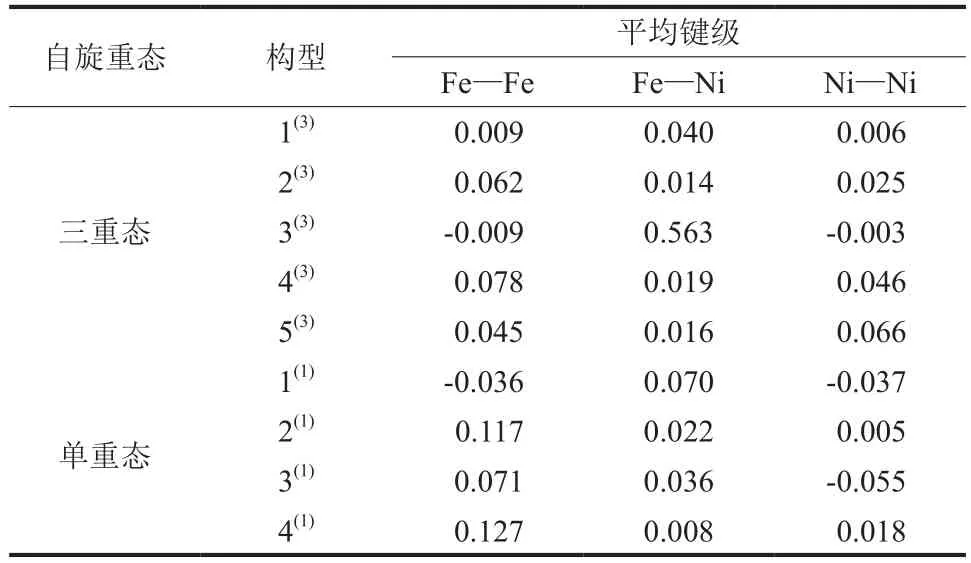
表2 团簇Fe3Ni3优化构型的平均键级Table 2 Average bond levels of optimized configurations of cluster Fe3Ni3
为了更为直观地分析Fe—Fe键、Fe—Ni键和Ni—Ni键的平均键级对构型稳定性的影响,将不同重态下各构型的各原子间键级平均值分别比该原子间的平均键级总值,即各成键键级在相应重态中的键级贡献比,计算时将平均键级为负值的键级设为0,将其计算结果绘制成图3。
由图3可知,单重态中,键级贡献比最大的是Fe—Fe键(57.81%);Fe—Ni键级贡献比为38.36%;而Ni—Ni键级贡献比最小,仅为3.83%。三重态中,Fe—Ni键级贡献比最大(41.75%);Fe—Fe键级贡献比为33.40%,与Fe—Ni仅相差8.35%;而Ni—Ni键级贡献比最小(24.85%),但与Fe—Fe键级相差8.55%,说明三重态中3 种成键键级对构型稳定性影响相差较小。值得注意的是,三重态中各成键键级贡献比相差较小(σ= 8.45),而单重态中各成键键级相差较大(σ= 27.34),再次证明自旋多重度对构型成键性质有影响。
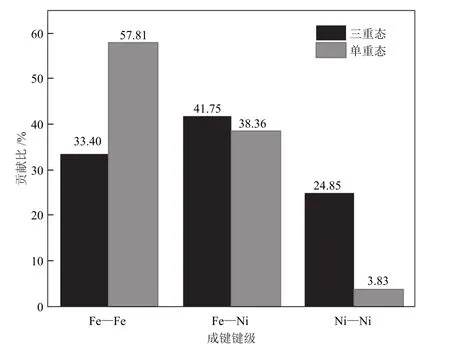
图3 团簇Fe3Ni3各类键级在单、三重态构型中的贡献比Fig.3 Contribution ratios of various bond orders in singlet and triplet configurations of cluster Fe3Ni3
综上,单重态中Fe—Fe成键键级对构型稳定性贡献最大,三重态中贡献最大的是Fe—Ni成键键级;Ni—Ni成键键级在单、三重态中贡献均最小,但在三重态构型中贡献远大于单重态构型,说明多重态影响各构型不同原子间成键,进而影响其稳定性。
3 结论
本文利用B3LYP/Lan12dz量子化学水平,在满足其计算精度的前提下,使用Gaussian09软件和Multiwfn软件进行计算,从态密度、原子间成键的键长、键级及键级贡献比等方面分析了团簇Fe3Ni3各构型的轨道杂化、成键性质,得到以下结论。
(1)团簇Fe3Ni3共有9种优化稳定构型(三重态构型5种,单重态构型4种),且三重态构型均较单重态构型稳定。各构型内部主要存在4s-4s、4p-4p、4p-3d与 3d-3d 4 种轨道杂化方式,其中 4s-4s轨道杂化作用最弱,对构型稳定性贡献最小,3d-3d轨道杂化的作用最强,贡献最大。但构型3(1)的轨道杂化主要由4p-4p与4s-4s两种方式组成,与其余构型均不同,值得对其构型空间结构中各原子间相互影响其轨道杂化方式进行更加深入的探究。
(2)Fe—Fe键在单重态下成键强度普遍高于三重态。三重态中Fe—Fe键与Fe—Ni键的存在对构型稳定性方面有促进作用,Fe—Ni成键键级对构型稳定性贡献最大;单重态中Fe—Fe键与Ni—Ni键的存在对构型稳定性具有促进作用,Fe—Fe成键键级对构型稳定性贡献最大,而Fe—Ni键与Fe—Fe键、Ni—Ni键之间存在拮抗作用,对构型稳定性产生一定程度的削弱。Ni—Ni键在三重态构型中的贡献远大于单重态构型,说明多重态影响各构型不同原子间成键,进而影响其稳定性。
本研究结论为该体系催化剂的制备提供了一定的理论指引,有助于选择最佳空间构型及成键方式,为新型催化材料的应用提供理论支撑。
