与微反应器一体化的Fe系催化剂制备及其费托合成催化性能研究
2022-12-27王彦谦任世杰王远洋
王彦谦,任世杰,王远洋
(太原科技大学 化学工程与技术学院,煤矸石高值利用山西省重点实验室,山西 太原 030021)
1923年,德国化学家Fischer和Tropsch开发了以CO和H2为原料,常压下催化合成液态烃类化合物的工艺,该工艺被命名为费托合成(Fischer-Tropsch synthesis)[1]。费托合成通常在固定床、浆态床和流化床这3类反应器上进行,这些反应器各有优劣:固定床的催化剂与产物易于分离,但催化剂床层可能出现局部飞温甚至烧结,传热和传质性能对反应进程的影响较大;浆态床传热和传质性能良好,催化剂床层压降也较小,但催化剂与重质烃类的分离比较困难;流化床产能大、传热效率高、催化剂可及时再生,但投资较高、催化剂损失较大且旋风分离器易堵塞,还极易因高温反应导致催化剂积炭和烧结。综上可见,这3类反应器均无法同时解决费托合成中的传热传质问题,以及催化剂与反应物/产物的分离问题。而在微反应器中,反应可在接近等温和低压降条件下进行,从而有望提高催化剂的活性和选择性,改善费托合成的反应效果[2]。
英国康帕克特GTL有限公司的C∙D∙李-塔夫内尔发明了一种采用机械加工方式制造的、波面或褶状蝶形和平片交替堆积的多条钢合金通道的费托合成紧凑型催化反应器,并通过在无孔钢合金通道基体上涂覆掺入催化材料的涂层,实现了生产力和选择性的良好平衡[3];大连理工大学的刘颖雅等[4]在微反应器金属基底上刻蚀微通道反应区域及微流体输入和输出通道,再通过酸碱处理获得活化金属表面,最后浸入含有机配体与金属盐的溶液进行催化剂原位制备,所得微反应器连续反应50 h仍可保持稳定的对有机污水的降解性能;中国石油化工股份有限公司的唐晓津等[5-9]采用酸蚀、激光刻蚀等方法制作平行线型结构或网状交叉结构的费托合成反应微反应器,分流构件为矩形或菱形,然后在微通道表面涂覆纳米级铁、钴、镍和钌等一种或多种催化剂层,或将以上纳米金属掺入液相介质中进行制备,所得微反应器具有体积小、结构紧凑,具有良好的移动性和投资低等特点;牛津催化剂有限公司的F∙达力等[10]在微反应器内填装催化剂颗粒进行费托合成反应,改进了催化活性和选择性。以上文献报道的费托合成微反应器的结构及其制作方法均较复杂,涉及机械加工、酸碱腐蚀和激光刻蚀等繁琐工艺或需填装催化剂颗粒,不利于推广应用。本课题组曾报道了一种在常规固定床反应器中填装微结构钢管,构筑微反应器的简便方法,以微结构钢管为Fe系催化剂基体,采用氢氧化钾腐蚀钢管表面后负载活性组分制成一体化催化剂,应用于费托合成反应,取得了良好效果[11]。
本文通过正交试验设计,在优化条件制备的微结构钢管(Fe)基体上,浸渍共活性成分(Mn、Co或Ce)制备了Fe系双组分催化剂,从而与钢管基体形成了一体化微反应器,并分别填装进常规反应器进行费托合成的催化性能评价,以筛选具有最优C2~C4烃类选择性的催化剂的制备参数,并采用扫描电镜(SEM)和X射线衍射(XRD)等对催化剂的形貌和结构进行表征,以期为该新型微反应器在费托合成中的实际应用奠定基础。
1 实验部分
1.1 实验试剂
本文所用的实验试剂见表1。

表1 实验试剂Table 1 Experimental reagents
1.2 与微反应器一体化的Fe系双组分催化剂制备
以优化条件制备的微结构钢管(碳20材质、每 5 根钢管熔融 20 g的KOH、腐蚀温度 500 °C,腐蚀时间3 h)作为基体(以下简称“钢管基体”),设计4因素3水平正交试验(表2)制备催化剂。催化剂制备步骤为:称取一定量Mn(NO3)2、Co(NO3)2或Ce(NO3)3共活性组分前驱体,移入烧杯中加蒸馏水搅拌至充分溶解达设定浓度(物质的量分数,下同);将钢管基体置于培养皿中,加入不同浓度的前驱体溶液,浸渍设定时间,每15 min翻动一次确保浸渍均匀;随后在室温下自然干燥后移入烘箱中,于 90 °C干燥 2 h,再移入马弗炉中,以 5 °C/min速率升温至设定温度,焙烧5 h后冷却至室温即得Fe系双组分催化剂,亦即共活性成分与钢管基体形成了一体化微反应器。根据表3中对活性组分、前驱体浓度、浸渍时间和焙烧温度4种因素和对应3种水平的约定,将所得催化剂记作样品D1~D9,将钢管基体记作样品D0,作为空白对照。
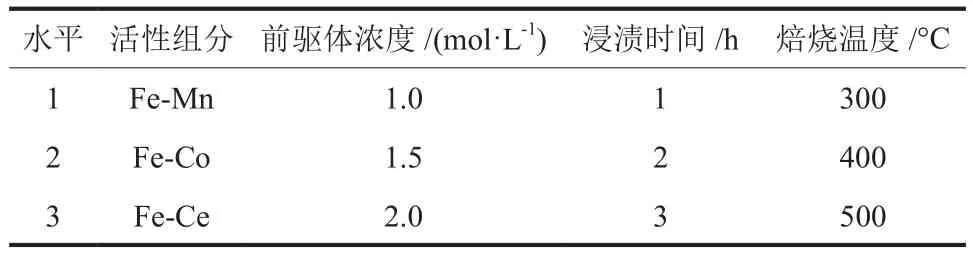
表2 制备Fe系双组分催化剂的正交试验设计Table 2 Orthogonal experimental design for preparation of Fe-based two-component catalysts
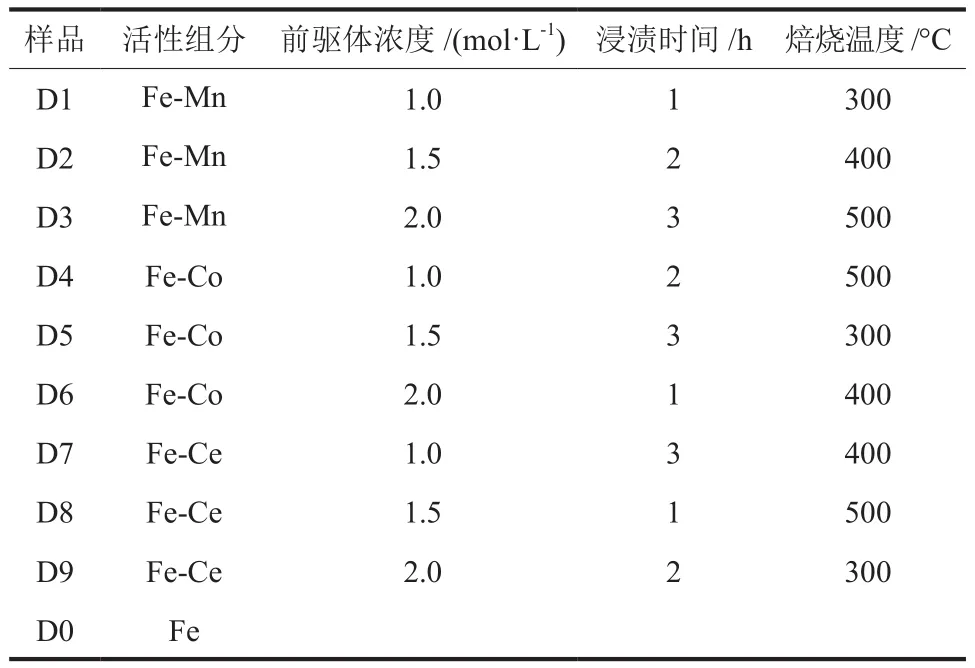
表3 Fe系双组分催化剂的制备条件Table 3 Preparation conditions of Fe-based two-component catalysts
1.3 催化性能评价方法
将制备好的样品装填进常规反应器中用于催化费托合成(图1),工艺条件为:反应温度300 °C、进料比V(H2):V(CO) = 2:3、空速 4000 h-1、反应压力0.1 MPa。CO和H2分别由质量流量计控制其比例和流速,经气体混合器混合后进入微反应器,由控温仪控制程序升温并反应,产物经背压阀和冷阱进入海欣GC-950型气相色谱仪,在线检测(一路通过TDX-01色谱柱采用热导检测器,另一路通过GDX-403色谱柱采用氢火焰离子化检测器)反应前后的气体组成。

图1 费托合成的微反应器装置Fig.1 Microreactor device for Fischer-Tropsch synthesis
采用专用工作站处理数据,以归一化法计算反应物转化率和各产物选择性,计算式见式(1)~式(5):

式中,XCO和XH2分别为CO和H2的转化率,%;SCO2、SCn和SC5+分别为CO2、轻烃(Cn,n= 1,2,3,4)和重烃(C5+烃类)的选择性,%;FCO,in和FH2,in分别为CO和H2进气量,mol;FCO,out、FH2,out、FCO2,out和FCn,out分别为CO、H2、CO2和轻烃出气量,mol。
1.4 催化剂表征方法
将催化剂样品截取至合适尺寸后粘贴在样品台的导电胶带上,采用日本株式会社JSM-7001F型扫描电镜,在不同分辨率下观察其微观形貌。采用日本理学株式会社MiniFlex 600型X-射线衍射仪表征催化剂样品的晶体结构,仪器参数为:Cu靶,λ= 0.1542 nm,工作电压 40 kV,扫描范围 5°~80°。
2 结果与讨论
2.1 反应温度对样品催化费托合成性能的影响
在进料比V(H2):V(CO) = 2:3、空速 4000 h-1和反应压力 0.1 MPa的条件下,分析了反应温度(220~340 °C)对样品催化费托合成性能的影响,结果见表4和表5。
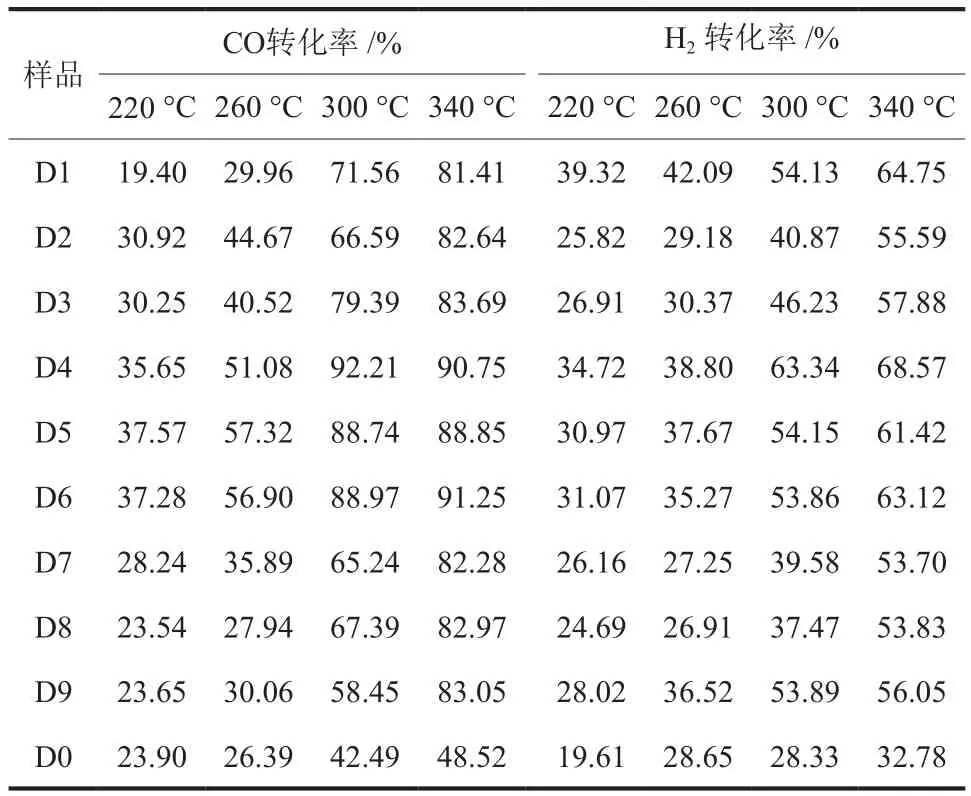
表4 反应温度对CO和H2转化率的影响Table 4 Effects of reaction temperature on CO and H2 conversion rate
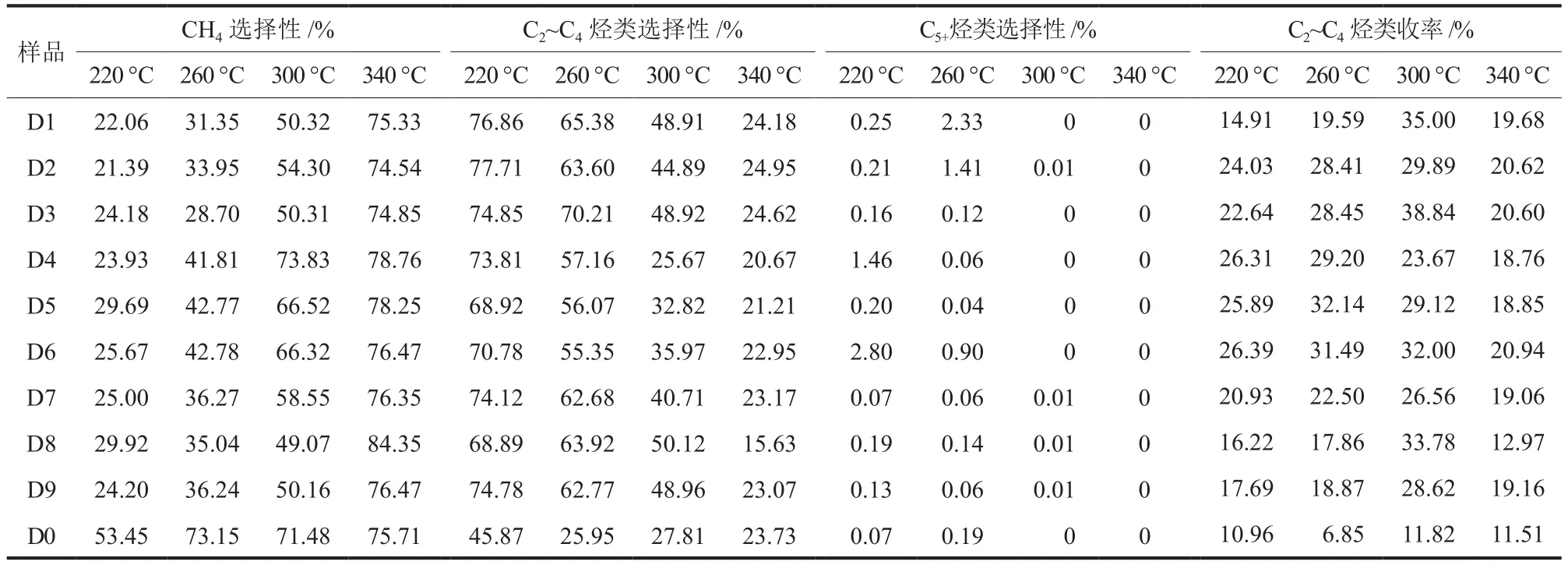
表5 反应温度对产物选择性和C2~C4烃类收率的影响Table 5 Effects of reaction temperature on products selectivity and C2~C4 hydrocarbons yield
从表4可以看出,随反应温度升高,所有样品的CO和H2转化率均提高,表明高温有利于CO和H2转化;对CO转化率而言,在所有反应温度下,Fe-Co双组分催化剂最高(D4~D6),Fe-Mn双组分催化剂次之(D1~D3),Fe-Ce双组分催化剂最低(D7~D9);对H2转化率而言则略有不同,D1明显高于D2和D3,仅次于D4而高于D5和D6,D9明显优于D7和D8,甚至优于D3和D2;值得注意的是,D1有利于H2转化而不利于CO转化,其原因有待进一步研究。
从表5可以看出,随反应温度升高,所有样品的C2~C4烃类选择性均下降,表明低温有利于降低CH4选择性并提高C2~C4烃类选择性,其中D1~D3最高,D7~D9次之,D4~D6最低;C5+烃类选择性整体很低,高于260 °C基本检测不到C5+烃类生成,在220 °C下,D6 和D4 最高(2.80%和 1.46%),在 260 °C下,D1和D2次之(2.33%和 1.41%),而在 220 °C和260 °C下,D7~D9均低于0.2%;同时检测出痕量二甲醚,可能的原因在于:根据表面烯醇机理,两个羟亚甲基缩合消除水形成了含有C—C键的二甲醚[12],表明二甲醚可能为生成C2及以上烃类的中间物种。C2~C4烃类收率为CO转化率与C2~C4烃类选择性的乘积,其值随反应温度升高先上升后下降,除D4外的所有样品均在300 °C达峰值,其中D3最高,达38.84%。
从表4和表5还可看出,作为对照的样品D0的CO转化率和C2~C4烃类选择性均较差,远低于D1~D9。
2.2 制备条件对C2~C4烃类选择性的影响
以反应温度300 °C的C2~C4烃类选择性为指标进行正交试验设计的数据分析(见表6),采用综合平衡法考察因素主次和水平优劣[13],表6中Ki值为各因素同水平之和,ki值为各因素同水平的平均值,由其大小可判断各因素的优水平和优组合;R值为各因素的极差,反映了各因素在水平波动时指标的变动幅度,R值越大,表明该因素对指标的影响越大,根据R值大小可判断因素的主次顺序。通过分析ki值得最优制备条件:活性组份为Fe-Mn、前驱体浓度为2.0 mol/L、浸渍时间为1 h和焙烧温度为300 °C;同时根据R值得出:活性组份是影响C2~C4烃类选择性的主要因素,前驱体浓度和浸渍时间的影响次之,焙烧温度影响最小。

表6 制备条件对300 °C下C2~C4烃类选择性的影响Table 6 Effects of preparation conditions on C2~C4 hydrocarbons selectivity at 300 °C
从表6可以看出,活性组份对C2~C4烃类选择性的影响为:D1~D3和D7~D9的C2~C4烃类选择性显著优于D4~D6,D1~D3略高于D7~D9,因此活性组分以Fe-Mn为宜。Co和Mn常用于制备费托合成催化剂,此处不再赘述,仅讨论Ce。BARTHOLOMEW[14]认为稀土金属氧化物可修饰Ⅷ族金属催化剂晶体表面,从而有利于CO解离,还可降低催化剂载体酸性(与K类似),抑制副反应(如烯烃裂解生成甲烷及其它轻烃等)发生;郭琪等[15]认为Ce的添加能够改善催化剂的还原性能,促进CO及H2在催化剂表面的吸附,从而影响催化剂的催化效果。前驱体浓度对C2~C4烃类选择性的影响是:随前驱体浓度增加,D4~D6的C2~C4烃类选择性逐渐增大,D1~D3和D7~D9也基本符合增大趋势,D1较高的选择性与其较短的浸渍时间(1 h)和较低的焙烧温度(300 °C)有关,而D9较低的选择性则与其较长的浸渍时间(2 h)有关,因此前驱体浓度以 2 mol/L为宜。浸渍时间对C2~C4烃类选择性的影响结果是:D7~D9和D4~D6 中,浸渍 1 h的D8和D6的C2~C4烃类选择性分别达到最大值,D1~D3 中,浸渍 1 h的D1 与浸渍 3 h的D3仅相差0.01%,因此浸渍时间以1 h为宜。焙烧温度对C2~C4烃类选择性的影响是:300 °C焙烧的C2~C4烃类选择性略高于 400 °C和 500 °C,由正交分析可知,焙烧温度对C2~C4烃类选择性影响最小,同时低温更有利节约能源,因此焙烧温度以300 ℃为宜。
2.3 催化剂表征分析
2.3.1 SEM分析
由于样品D3的C2~C4烃类收率最高(表5),对其表面形貌进行了SEM分析(图2(d)~(f)),以样品D0作为对比(图2(a)~(c))。从SEM照片中可以看出,样品D0表面凹凸不平,存在很多不规则微坑(图2(a)),微坑表面粗糙且存在大量孔隙(图2(b)),同时残留少量白色KOH(图2(c))。事实上,样品D0的粗糙表面有利于共活性成份的附着;浸渍的共活性成份又可将样品D0的粗糙表面“填平补齐”,从而使样品的表面趋于平滑。从图2(d)~(f)可以看出,浸渍Mn后,样品D3表面仅有少量不规则微坑和孔隙,说明其表面活性组分分布相对均匀,进而对费托合成起到催化促进作用。
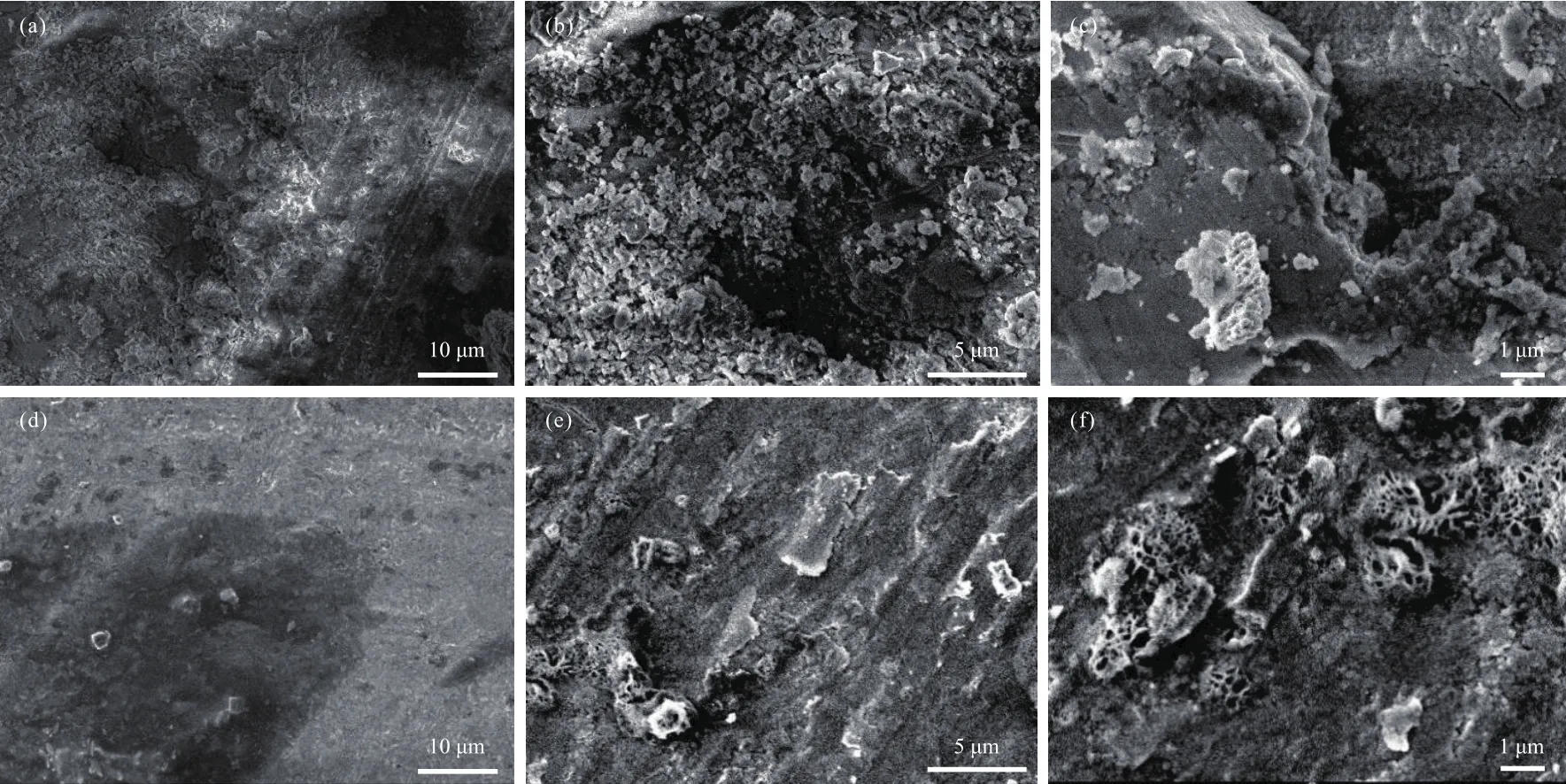
图2 样品D0((a)~(c))和样品D3((d)~(f))的SEM照片Fig.2 SEM images of sample D0 ((a)~(c)) and sample D3 ((d)~(f))
2.3.2 XRD分析
图3为D1~D9样品的XRD谱图。从图3可以看 出,所 有 样 品 在 24.2°、33.1°、35.7°、40.8°、49.5°、54.9°、62.4°和 63.9°处的衍射峰归属于Fe2O3;D1~D3在 37.3°和 56.2°处的衍射峰归属于MnO2,29.5°处的衍射峰归属于MnFe2O4;D4~D6 在 31.2°、36.8°、44.7°和 59.3°处的衍射峰归属于Co3O4;D7~D9 在28.5°、47.4°和 56.4°处的衍射峰归属于CeO2,均与文献报道的衍射峰位置相同[16-18],说明Mn、Co和Ce均已负载至钢管基体上,D3、D6和D9的衍射峰高度分别为同类样品中最高,表明其晶化程度比同类样品更高,这显然与其高前驱体浓度有关。

图3 不同样品的XRD谱图Fig.3 XRD patterns of different samples
3 结论
以优化制备的微结构钢管(样品D0)作为基体和对照,通过正交试验设计,采用浸渍法制备了Fe-Mn、Fe-Co和Fe-Ce双组份催化剂(分别对应样品D1~D3、D4~D6 和D7~D9),从而与基体形成一体化微反应器,并对其催化费托合成反应的性能进行了考察,得出以下结论。
(1)高温有利于提高CO和H2转化率,而低温有利于降低CH4选择性并提高C2~C4烃类选择性,C2~C4烃类收率几乎均在300 °C达峰值,均显著高于D0,其中D3最高,达38.84%。
(2)以反应温度300 °C的C2~C4烃类选择性为指标进行正交分析,得最优制备条件为:Fe-Mn活性组份、2.0 mol/L前驱体浓度、1 h浸渍时间和 300 ℃焙烧温度。活性组份是影响C2~C4选择性的主要因素,前驱体浓度和浸渍时间的影响次之,焙烧温度影响最小。
(3)浸渍共活性组份后样品表面趋于平滑,仅有少量不规则微坑和孔隙,其相对均匀的表面活性组份分布有助于促进对费托合成的催化性能;Mn、Co和Ce均已负载至钢管基体上,高前驱体浓度制备的样品的晶化程度更高。
