人工智能在新药研发中的应用进展
2022-12-23杜晗吴羿霏杜新
杜晗,吴羿霏,杜新
(1. 澜亭资本,浙江 杭州 310030;2. 深圳埃格林医药有限公司,广东 深圳 518038)
1 人工智能的概念
人工智能(artificial intelligence,AI)这一概念已不再陌生,其通常是指通过计算机程序或系统来呈现人类思维和智能的技术。AI通过抓取数据中存在的概念和关系,独立分析学习数据模式从而达到模拟人类思维的目的[1]。AI主要涉及的方法领域包括推理、知识表达、搜索解决方案以及机器学习(machine learning)[2]。
德勤咨询报告将机器学习归属于广义的AI技术,并定义为“从结构化和非结构化数据中学习、识别隐藏模式、进行分类并预测未来结果的计算机算法”。机器学习技术包括深度学习(deep learning),它是一种基于机器学习的方法,利用一种类似于大脑的逻辑结构(称为神经网络)来识别和区分语音、图像和视频等模式。自然语言处理(natural language processing,NLP)是深度学习的一种,是计算机技术在自然语言和语音分析与合成中的应用[1](见图1)。

图1 机器学习与深度学习的关系Figure 1 The relationship between machine learning and deep learning
深度学习是机器学习领域中一个新的研究方向,引领了第3次AI的浪潮。深度学习的算法近年来发展迅速,几种典型的算法,包括卷积神经网络(convolutional neural network,CNN)、循环神经网络(recurrent neural network,RNN)、深度强化学习(reinforcement deep learning),特别是生成对抗网络(generative adversarial network,GAN),作为最近2年十分热门的一种无监督学习算法,在各个领域被广泛应用。
2 人工智能在新药研发中的具体应用
新药研发是一个漫长而复杂的过程,伴随着高昂的研发成本及高度不确定性。有学者统计,一个新药从研发初期到上市,可能需要数十年,消耗资金可多达30亿美元。典型的新药研发过程通常包括:1)早期的目标识别及靶点、最优化合物的确认;2)临床前研究;3)临床研究Ⅰ、Ⅱ、Ⅲ期阶段;4)审批上市阶段。在化学世界里,潜在药物分子多达1060个,因此新药发现可以说如同大海捞针[3]。近年来,传统的新药研发越来越难,研发投入和研发时间也在不断增加。新药研发处于一个难以突破的瓶颈阶段,亟需借助于新的技术来帮助实现降本增效。
AI凭借其强大的自适应特征和学习能力,将其算法、推演等核心技术应用到新药研发的各个环节,在保证分析质量的同时,大幅降低药物研发成本,缩短研发时间,提高研发效率,使新药开发走上快速高效的道路。
Paul等[2]于2021年在Drug Discovery Today期刊上发表了一篇文章,总结了AI技术在新药研发的5个重要部分——药物设计、化学合成、药物再利用、多重药理学、药物筛选中的巨大潜力(见图2)。以下从这5个领域进行详细介绍。
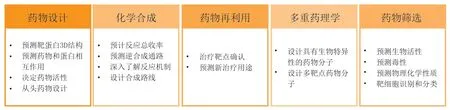
图2 人工智能技术在新药研发中的应用潜力Figure 2 Application potential of artificial intelligence technique in new drug research and development
2.1 药物设计
2.1.1 预测靶点蛋白三维结构蛋白质功能紊乱是导致许多疾病的重要因素,因此在新药研发第一步——靶点识别和确认中,找到药物在体内的作用靶点,确定靶点蛋白质结构从而针对性地设计药物分子来调节蛋白功能是至关重要的基石。蛋白质的三维结构由一系列氨基酸折叠而成,根据最稳态原理推测出蛋白质三维结构需要考虑到其各个原子间的相互作用力,因此巨大的计算量使得蛋白质结构测定往往需要很长的周期和高昂的经费,且难度较大。
到目前为止,在生命体已知的约2亿种蛋白质中,只有约17万种蛋白质结构得到了确定。基于已知的氨基酸序列,AI被研究人员用于预测蛋白质结构。Spencer等[4]在2015年通过深度学习网络架构(deep learning network architecture,DNSS)对198个蛋白质进行完全独立测试,预测蛋白质二级结构准确率达到了80.7%。Wang等[5]在2016年发表于《自然》的文献里介绍到一种被称为深度卷积神经场(deep convolutional neural fields,DeepCNF)的AI技术,其被用于预测蛋白质二级结构。DeepCNF技术不仅可以通过深度层次结构对复杂的序列与结构关系建模,还可以对相邻的蛋白质二级结构之间的相互依赖性进行建模。实验结果证明该技术可实现84%的预测准确率,并可以扩展到对蛋白质的其他结构性质(如接触数、无序区域以及溶剂可及性)的预测。2020年11月,谷歌旗下的DeepMind公司的AlphaFold AI系统在国际蛋白质结构预测竞赛(Critical Assessment of Structure Prediction,CASP)上以绝对的优势夺冠,其预测的蛋白质三维结构与实验方法解析的结构几乎完全吻合,在最具挑战的蛋白质结构预测上也获得将近90分的成绩,而其他参赛队伍最好的成绩也只有75分[6]。由此可见,AI技术在实现蛋白质结构的精准预测上有巨大潜力,将会为新药研发带来前所未有的技术革新。
2.1.2 预测药物和蛋白质相互作用确定药物的靶蛋白,以及预测药物和靶蛋白的相互作用(drug-protein interaction,DPI),在药物研发过程中有着极其重要的作用。通过预测药物与受体或者蛋白质的相互作用,可以帮助理解药物的功效,从而对药物进行最有效的设计。
得益于愈发丰富的数据库(例如化合物数据库PubChem、ChEMBL与DUD-E,蛋白质数据库UniProt与PDBbind,以及综合数据库BindingDB),许多研究人员采用了不同的AI技术对配基-蛋白质的相互作用成功地进行了预测。Wang等[7]采用支持向量机(support vector machine,SVM,一种按监督学习的方式对数据进行二向分类的广义线性分类器)对存在于626个蛋白质和10 000个活跃化合物之间的15 000个配基-蛋白质相互作用进行训练,成功地发现了9种新化合物及其与4个关键靶点(GPR4、SIRT1、p38和GSK-3β)的相互作用。
药物分子与细胞网络中蛋白质的相互作用对于药物开发非常重要,然而,现有的识别药物-蛋白质相互作用的预测器都是由一个偏态分布的基准数据训练的,在这种偏态分布的基准数据中,非相互作用的药物-蛋白质对的数量远远大于有相互作用的药物-蛋白质对。使用这种高度不平衡的基准数据来训练预测器将导致许多有相互作用的药物-蛋白质对可能被错误地预测为没有相互作用的结果。由于少数有相互作用的药物-蛋白质对通常包含药物设计最重要的信息,因此有必要尽量减少使用这种偏态分布的基准数据,以免造成错误预测。Xiao等[8]采用邻域清理规则和合成少数过量采样(synthetic minority over-sampling)技术来处理偏态分布的基准数据,由此获得新的优化基准数据,在此基础上开发了一个名为iDrug-Target的新预测器,其中包含4个子预测器:iDrug-GPCR、iDrug-Chl、iDrug-Ezy和iDrug-NR,专门分别用于鉴定药物分子与G蛋白偶联受体(GPCR)、离子通道、酶和核受体(NR)的相互作用。对一组实验确认的数据集进行的严格的交叉验证表明,这个新的预测器在相同的目的上明显优于现有的其他预测器。
药物设计从早期的计算机辅助药物设计逐步发展为AI辅助药物设计,随着AI计算能力的不断加强、数据库数据的增加以及数据处理能力的优化,AI辅助药物设计的能力和范围也大大增加。
2.2 化学合成
药物发现阶段的复杂性是导致新药研发高成本的重要原因之一。采用先进的计算机硬件和计算方法优化药物化学在新药研发设计-制造-测试-分析(design-make-test-analyze,DMTA)中的表现,将提高新药研发的成功率。其中,研究人员越来越感兴趣的一个领域是使用数据驱动的合成预测工具使合成阶段加速以及减少新分子实体合成的失败。
计算机辅助化学合成综合规划包括以下3个主要任务。1)逆合成。逆合成又可以分为2个方面:对逆合成的每一步合成提出建议;递归使用单步建议来识别完整的多步路线。2)提供正向反应的条件,从而使提供的建议具有可操作性。3)反应预测。从一组起始原料和条件预测可能的产物,用于验证所提出的合成步骤[9]。
计算机辅助化学合成可以追溯到20世纪60年代,在60至90年代,该学科在很大程度上受限于计算资源,主要依赖于人工编码。近年来,AI专家尝试让用于预测的机器学习模型与化学家在化学合成方面进行密切“合作”,旨在建立更加快速、方便和简单的服务平台。随着机器学习的不断发展,化学家们不断接受使用计算机辅助化学合成来减轻他们的工作量,以及应对日益增长的合成挑战。
美国麻省理工大学在其药物发现和合成的机器学习联盟(Machine Learning for Pharmaceutical Discovery and Synthesis Consortium,MLPDS)的平台上,基于麻省理工大学的化学工程、化学和计算机科学家与拜耳、辉瑞、诺华等多家大型跨国医药公司的合作,共同设计用于化学分子发现和化学合成自动化的应用软件,以帮助医药公司加速化学药物的发现、合成和制造。大学和大型医药公司间的通力合作将有利于AI在化学合成领域更深入和广泛的应用。
2.3 药物再利用
药物再利用[或药物再定位(drug repurposing)],是指将已上市的药物,以及正在进行研究的药物和临床失败的药物,用于原定用途之外的疾病治疗的过程。正如诺贝尔奖获得者、药理学家James Black的一句名言所说,发现新药最富有成效的基础就是老药[10]。得益于老药已知的安全性,药物再利用不仅可以大幅度降低研发成本,还可以有效减少药物安全性测试的相关风险,因此也是新药研发中比较重要的一种研发策略[11]。
如今,AI技术已经被广泛用于药物再利用的系统研发过程中。Rodriguez等[12]开发了机器学习框架——DRIAD(drug repurposing in Alzheimer’s disease),其能够量化药物作用与阿尔茨海默病进展之间的关联。该研究团队用80种测试化合物(主要为具有抗肿瘤活性的激酶抑制剂,有33种已获FDA批准)体外处理人类神经细胞,收集受到影响的基因并形成列表。将DRIAD用于分析该基因列表,从而得到一个可能再利用的候选药物的排名表。对列表中得分最高的药物进行检查,有助于了解其靶点的共同特点。该AI框架能够提供直接、客观的量化结果。
Zhou等[13]在《柳叶刀》上发表的一篇文献总结了不同AI技术用于药物再利用的研究,其中探讨了将老药用于新冠肺炎(COVID-19)治疗的可能性。这些技术包括前馈神经网络(feedforward neural network)、主要分析处理图像输入变量的CNN、更适合处理生物序列的RNN以及图表表征学习(graph representation learning)。通过药物网络分析发现了或可用于新冠肺炎治疗的候选药物托瑞米芬(toremifene),该药曾于1997年获批准用于治疗乳腺癌,为第1代雌激素受体调节剂。体外实验表明,托瑞米芬在微摩尔浓度下可阻断病毒感染,包括中东呼吸综合征冠状病毒、新冠病毒(SARSCoV-2)。经过Benevolent AI公司的知识网络图谱分析与识别,巴瑞替尼(baricitinib)也被认为对新冠肺炎有一定疗效。目前,至少2项以巴瑞替尼(单独使用或与抗病毒药物联合使用)治疗中度和重度新冠肺炎患者的双盲Ⅱ期临床试验正在美国进行。
AI辅助药物再利用可以加速药物开发进程,减少开发成本。除了新冠肺炎外,AI辅助药物再利用在其他适应证上也可以得到广泛应用。已知药物拥有庞大的临床和毒理数据,AI利用其强大的计算能力和算法在寻找新的适应证和新的药物组合中都可以发挥重要作用。
2.4 多重药理学
在现代药物研发领域,大多数治疗方法都是通过调节多个靶标和通路来达到预期效果,而不是通过作用于单个靶点起效。多重药理学(polypharmacology)作为制药科学的一个新兴分支,着重研究两个方面:1)由药物分子脱靶效应导致的毒副作用;2)利用药物分子同时调节疾病网络系统中的多个靶点,从而治疗复杂疾病。由于要处理的数据量很大,网络药理学(network pharmacology)、机器学习技术和化学基因组学方法在该领域的应用是必不可少的。根据美国国家医学图书馆的定义,多重药理学是“对作用于多个靶点或途径的药物的设计或使用”。癌症、阿尔茨海默病等诸多复杂疾病通常在环境、遗传等多种因素的共用作用下引起,而多重药理学已成为一种强大和有前途的药物开发手段,以满足迫切的医疗需求。
在过去,多靶点药物的发现是偶然的。随着化学生物学和计算机科学的发展,基于多重药理学的药物合理设计成为可能。现代体外高通量/高含量筛选和体内动物模型技术加快了对药物靶点组合的系统识别,而在电脑模拟(in silico)方法中,结构生物学和药物化学使多靶点药物的有效设计得以实现。
基于配体的多重药理学预测方法主要依赖于药物的化学结构和生物活性。最近,Lee等[14]利用1 121个靶点,通过AI随机森林方法开发了基于配体的靶点预测模型。结果显示,对于前1%和前3%的目标,这两组目标的回忆率分别达到了67.6%和73.9%。
总部位于加拿大多伦多的生物科技公司Cyclica采用多重药理学方法进行药物发现,将药物分子所有潜在靶标的相互作用作为首要考虑因素。Cyclica的集成AI增强药物发现平台能够对具有良好多重药理特征和药用特性的候选药物进行多目标评估和设计。传统计算机辅助药物设计方法(如基于结构的生物物理学方法)需要在有大量数据存在的分子类别和蛋白质靶标上才能发挥最大作用,而Cyclica的平台可针对特征不太明确的蛋白质靶标进行分子设计,同时也揭示了分子的作用机制。Cyclica平台将目标反褶积应用于药物再利用和重新设计。该平台由2个机器学习引擎——MatchMaker和POEM提供动力[15]。
2.5 药物筛选
虚拟筛选(virtual screening)是计算机辅助药物设计中一种重要的开发工具,利用小分子化合物与药物靶点间的分子对接运算,从庞大的分子库中快速遴选出活性化合物。传统的高通量筛选技术存在高成本、低成功率的问题,虚拟筛选则可通过在早期药物研发时过滤掉结构不合适的化合物,提高化合物筛选效率,从而缩短研发周期,降低药物研发成本。
虚拟筛选主要的2种方法有基于配体的虚拟筛选(ligand-based virtual screening,LBVS)与基于结构的虚拟筛选(structure-based virtual screening,SBVS)。前者不依赖于三维蛋白质结构信息,而是基于活性及非活性配体的实证数据,利用活性配体之间的化学和空间相似性及物理化学分析来预测和识别其他具有高生物活性的配体[11]。传统的机器学习方法,例如SVM(support vector machine)、NB (Naïve Bayes)和KNN(k-nearest neighbour)被大量运用在LBVS中。而SBVS则依赖于实验测定的或同源模建的受体生物大分子的三维结构,主要用于研究可能的活性配体与结合位点残基间的相互作用。SBVS通过打分函数对蛋白和小分子化合物的结合能力进行评价,最终从大量的化合物分子中挑选出结合模式比较合理的、预测得分较高的化合物,往往比LBVS有更好的预测表现。
AI在药物筛选中还有多方面的应用,包括预测药物生物活性、药品物理化学性质和药物毒性。在预测药物生物活性方面,由于药物分子的生物活性取决于它们对靶蛋白或受体的亲和力,药物-靶点结合亲和力(drug-target binding affinity,DTBA)对于预测药物与靶点的相互作用至关重要。AI可以根据药物的特征或其与靶蛋白或受体相互作用来测量结合亲和力,从而预测药物生物活性。在这一方面,近年来许多AI工具,如DeepDTA、PADME、WideDTA和DeepAffinity均被用于测量药物与靶点的结合亲和力。相比而言,深度学习的方法因为使用基于网络的方法,可以不依赖蛋白质的三维结构,因此比机器学习的方法具有更好的预测性能。
测试药物分子的毒性对于药物开发至关重要。通常做法是先通过体外细胞试验进行初步研究,然后进行动物试验来确定毒性,这种传统的毒性测试方法费时而且成本较高。AI为药物分子的毒性测试提供了快速、低成本的途径,相关工具有LimTox、pkCSM、admetSAR和Toxtree。AI方法通常是通过比较药物分子的相似性,或通过输入药物分子的特性来预测其潜在毒性。Mayr等[16]使用AI工具DeepTox,通过识别药物分子的静态和动态分子特性,如药物分子的相对分子质量和范德华力,有效地预测了药物分子的毒性。
3 人工智能在实际药物研发中的成功案例
截 至2021年4月12日,据BiopharmaTrend.com网站统计,基于AI技术进行药物研发的公司已多达270家,其分散在10个不同领域,包括化学合成、靶点/先导化合物确认、临床前研究、药物再利用等。
AI公司与大型跨国药企的战略合作已屡见不鲜。葛兰素史克(GSK)于2017年同美国的Insilico Medicine达成合作协议,希望借助后者的AI平台发现新的药物靶点及通路。2017年6月,基因泰克(Genentech)宣布与GNS Healthcare进行研究合作,利用该公司自有的“逆向工程、正向模拟”(reverse engineering, forward simulation,REFS)机器学习和模拟平台去识别与验证新的癌症治疗药物靶点。武田制药也宣布同AI药物设计公司Numerate进行多年研究合作,专注开发肿瘤学、胃肠病学和中枢神经系统疾病的临床候选药物[17]。以下重点介绍AI公司Exscientia和Benevolent AI及其AI技术在药物研发中取得的一些成果。
3.1 Exscientia公司
Exscientia是一家位于英国牛津,通过端对端的AI平台对药物进行设计的制药科技公司,也是首家实现药物设计自动化的公司。许多大型跨国药企已经分别与Exscientia达成了战略合作。2017年,GSK与Exscientia签署协议,就GSK筛选的10个疾病靶点通过AI平台开发小分子药物,并且针对这些靶点发现临床候选药物,如果Exscientia能够实现所有的“里程碑”,则会获得GSK提供的3 300万英镑的研究资金[18]。同年,赛诺菲出资2.73亿美元,与Exscientia共同开发治疗糖尿病及其并发症的双特异小分子药物,涉及领域包括血糖控制、体质量管理及其他糖尿病相关领域。双方已经筛选了45个单靶点和1 000个双靶点组合,随后会借助Exscientia的AI平台剔除掉在化学上难以处理的组合。拜耳和Exscientia于2020年初签署了一份为期3年、价值2.4亿欧元的合作协议,通过结合Exscientia的AI药物研发平台以及拜耳的数据,专注药物早期研究,识别及优化具有心血管疾病和肿瘤治疗作用的先导化合物[19]。
许多与Exscientia合作的制药公司在新药研发方面已经取得了一些进展。2022年6月14日,Exscientia发布其与Evotec基于AI共同合作研发的抗肿瘤药物EXS21546在健康受试者中的Ⅰ期研究数据。该药物是一款腺苷A2A受体拮抗剂,用于治疗成人晚期实体瘤[20]。
3.2 Benevolent AI公司
Benevolent AI是一家成立于英国伦敦的独角兽公司,其主要利用AI技术,从各种结构化以及非结构化的生物医学数据源中提取数据并进行整理、归纳、标准化,将数据输入到公司的专利知识图谱中,从而提取出能够推进药物研发的信息,加速药物研发的过程。
阿斯利康与Benevolent AI于2019年达成战略合作,结合阿斯利康自有的基因组学、化学与临床数据,以及Benevolent AI靶点识别平台与生物医学知识图谱,对慢性肾病和特发性肺纤维化的相关治疗药物进行研发[21]。该合作在2021年1月27日取得了阶段性的成功,阿斯利康在Benevolent AI的AI技术帮助下,在导致慢性肾病的潜在机制中,成功发现了新的药物靶点,并将该靶点纳入了公司的新药开发计划[22]。
目前,Benevolent AI的药物研发项目主要涵盖过敏性皮炎、肌萎缩性脊髓侧索硬化症、溃疡性结肠炎、炎症性肠病、中枢神经系统疾病、胶质母细胞瘤、非酒精性脂肪肝,与阿斯利康合作的疾病领域有慢性肾病、特发性肺纤维化。通过Benevolent AI的AI平台研发的过敏性皮炎治疗药物BEN-2293已进入Ⅰ期临床试验,该药是一款多靶点小分子pan-Trk拮抗剂,也是一款“best-in-class”创新药[23]。
4 人工智能应用于药物发现的挑战与思考
尽管AI在药物发现方面展示出巨大的前景,但其也面临着数据缺失、缺乏可解释性等诸多挑战。
在所有涉及AI的行业中,缺乏数据是一个反复出现的问题。在传统的生物学研究中,有效样本的数量有限,而大多数机器学习算法必须在成百上千的数据点或样本上进行训练才能表现良好。另一个挑战是缺乏可解释性——通常很难解释模型如何进行某些预测和执行。缺乏可解释性更可能发生在深度学习中,其中每一层都增加了模型的复杂性。随着层数的增加,对每一层输出的解释其复杂程度可能以指数级增长。
使用AI预测药物靶点的一个难点是如何将全球实验室进行的传统基础研究翻译成计算机可以理解的语言。机器学习程序依赖于既可以识别模式又可以训练机器的数据,这通常需要复杂的实验设计,将人为错误保持在最低限度,并且可以在几乎相同的条件下执行多次不同的实验迭代。
与任何给我们对现有技术的理解带来转变的进步一样,AI目前仍然无法在药物发现过程中完全取代人类科学家。计算机的预测必须经过科学家的验证,以确保AI在药物研发中的有效性和实用性。
5 结语与展望
AI在新药研发中有着巨大的应用前景,其中部分技术已取得了阶段性的进展,基于AI研发的一些新药已进入临床研究,不少AI医药研发公司也在不断取得新的成果。但是,AI技术在研发中若要取得井喷性和长期有效的成果,还有很长的路要走。
AI+新药研发是一项跨学科、跨行业的合作,极需精通AI技术及生物制药技术的复合型人才,也需要IT行业与医药行业,高校、科研院所与医药公司的通力合作。跨国药企可以通过其雄厚的研发力量在该领域取得成果。中小型的药企也可以找准目标,确定突破口,同时积累人才,做好转型工作,从而快速取得成果。
AI+新药研发要取得持久的研发成果和商业价值,也需要医药公司与投资行业的紧密合作。只有大规模的投资进入这一领域,营造出良好的行业状态和环境,才能使药物从初期探索顺利走向临床、最终进入商业市场,造福广大患者的同时,也为研发者和投资者带来回报。
