THI3/BAT2基因缺失酿酒酵母菌构建及其在黄酒酿造中的应用
2022-12-14陈文颖周世水闫统帅梁思宇
陈文颖,周世水*,闫统帅,梁思宇
(1.华南理工大学生物科学与工程学院,广东广州 510006)(2.广东石湾酒厂集团股份有限公司,广东佛山 528031)
黄酒是我国的民族特产之一,风味独特,富含矿物质、维生素、有机酸、肽、氨基酸、γ-氨基丁酸、多酚等营养物质和生物活性成分[1]。但我国传统黄酒中高级醇含量较高,消费者普遍反映有易上头、易醉等现象[2]。高级醇,也称为杂醇,是酒精饮料中的一类风味化合物,对酒精饮料的感官品质和特性有显著的影响。适量的高级醇会使得酒体口感醇厚、柔软、丰满,酒香协调。当高级醇含量过高时,酒不仅会有杂醇油的味道,而且容易有“上头”的强烈醉感[3]。黄酒中高级醇对人体有潜在的危害,它比乙醇具有更高的毒副作用[4,5];其次不同高级醇比例导致毒副作用不同,特别是异戊醇/异丁醇比例越高,酒的醉度越高[6]。采取有效发酵控制来调控黄酒中高级醇的含量及不同高级醇比例,降低黄酒“上头”的醉度和改善黄酒品质,对提高黄酒的地位和扩大市场消费量具有重要意义。
酵母发酵产生的高级醇主要来源于糖代谢生物合成途径或支链氨基酸Ehrlich 降解途径[7,8],在Ehrlich途径中,BAT2基因编码的支链氨基酸转氨酶催化支链氨基酸转化为α-酮酸,随后进一步转化为高级醇[9]。THI3基因编码一个与丙酮酸脱羧酶(PDCs)十分相似的蛋白质类酮酸脱羧酶,被鉴定为亮氨酸代谢的主要脱羧酶,催化2-酮异己酸形成异戊醇[7]。目前BAT2基因[9-11]和THI3基因[12-14]单独缺失对不同酒类影响均有较多研究,但还对THI3和BAT2基因同时缺失对黄酒影响的研究还较少。
本文以酿酒酵母(Saccharomyces cerevisiae)菌株XF1 的单倍体XF1a7/XF1α6 和THI3基因缺失XF1-T的单倍体XF1a7-T/XF1α6-T 为原始菌采用Cre/loxP 同源重组系统[15]构建BAT2基因缺失重组菌XF1-B 和THI3/BAT2双基因缺失重组菌XF1-TB,然后进行黄酒发酵研究BAT2基因缺失和THI3/BAT2双基因缺失对高级醇生成的影响。以期控制黄酒中高级醇含量和优化高级醇比例来降低黄酒“上头”的醉度,为提高黄酒品质提供参考。
1 材料与方法
1.1 材料与试剂
1.1.1 菌株与质粒
本研究中使用的所有菌株和质粒载体列于表1。

表1 本实验的菌株和质粒Table 1 Strains and plasmids used in this study.
1.1.2 引物设计
本研究中使用的引物列于表2中。酿酒酵母(S288c)BAT2基因组序列从NCBI(序列号:NC-001144.5)获得。采用Snapgene 软件设计引物,上海生物工程技术有限公司合成。

表2 本实验使用的引物Table 2 Primers used in this study
1.1.3 主要试剂
中性蛋白酶、风味蛋白酶,河南万邦化工科技有限公司;ApexHFHS 脱氧核糖核酸聚合酶、DNA 回收纯化试剂盒、质粒小量提取试剂盒,湖南艾科瑞生物科技有限公司;遗传霉素(G418)和博来霉素(Zeocin),北京普博欣生物科技有限责任公司;氨基酸标准品、乙酸丁酯、丙酮、正丙醇、异丁醇、异戊醇、β-苯乙醇(均为色谱纯),上海阿拉丁生化科技股份有限公司;半乳糖、无水乙酸钠、氯化钾、氯化钠、氢氧化钠(均为分析纯),天津市科密欧化学试剂有限公司;异硫氰酸苯酯,上海阿拉丁生化科技股份有限公司;酵母提取粉、蛋白胨、琼脂(生化试剂):广东环凯微生物科技有限公司。
1.1.4 主要培养基
LB 培养基:氯化钠10 g/L、蛋白胨10 g/L、酵母提取粉5 g/L。
YPD 培养基:葡萄糖20 g/L、蛋白胨20 g/L、酵母提取粉10 g/L。
YPG 培养基:半乳糖20 g/L、蛋白胨20 g/L、酵母提取粉10 g/L。
McClary 麦氏产孢培养基[16]:葡萄糖1 g/L,氯化钾1.8 g/L,无水乙酸钠8.2 g/L,酵母提取粉2.5 g/L,琼脂20 g/L。
所有固体培养基在液体培养基的基础上添加20 g/L 琼脂,并均经121 ℃高压蒸汽灭菌20 min,备用。
1.2 仪器与设备
TC1000-S PCR 扩增仪、PowerPac 3000 电泳仪、GelDoc 2000 凝胶成像分析系统,美国Bio-Rad 公司;CanNeed-MB-800 自动糖化器,肇庆市嘉仪仪器有限公司;5804R 高速台式冷冻离心机,德国Eppendorf公司;GC8100 气相色谱仪,滕州中科普仪器有限公司;高效液相色谱仪,LC-10A 双泵岛津液相色谱;安捷伦TC-C18 色谱柱,安捷伦科技(中国)有限公司。
1.3 方法
1.3.1 敲除组件构建
以pUG6 质粒为模板,用引物loxP-F/loxP-R 进行PCR 扩增,获得两端与酵母基因组同源的KanMX抗性基因片段(1 736 bp);以提取的酵母基因组DNA 为模板,用引物BAT2-F1/BAT2-R1 和BAT2-F2/BAT2-R2 分别PCR 扩增出同源臂BU(290 bp)和BD(361 bp)。利用KanMX 基因两端与两同源臂的重叠碱基进行融合 PCR 连接成BAT2基因敲 除组件BU-loxp-KanMX-loxp-BD(2 338 bp)。
1.3.2 酿酒酵母电转化和阳性转化子筛选
电转化法[17]将敲除组件导入单倍体酿酒酵母细胞XF1a7/XF1α6 和XF1a7-T/ XF1α6-T,加0.1~0.2 μg纯化敲除组件DNA 到感受态细胞,电转化(1.5 kV、200 Ω、25 mF 的电容电击5 ms)后,温育的转化子涂布含100 μg/mL G418 抗生素的YPD 平板上培养。挑取阳性菌落用引物BAT2-A、BAT2-D 进行PCR 验证。
1.3.3 筛选标记去除与质粒丢失
利用电转化法将带有Cre 重组酶基因的pSH65 质粒导入BAT2基因缺失的单倍体酵母细胞XF1a7-BK/XF1α6-BK 和XF1a7-TBK/XF1α6-TBK,涂布于含200 μg/mL zeocin 抗生素平板上培养3 d,挑取阳性菌在YPG 液体培养基中连续培养10 h 以上,以期有足够的时间使半乳糖诱导质粒pSH65 产生Cre 重组酶,通过Cre 重组酶切除loxP位点间的抗性基因,然后涂布于含G418 平板和不含G418 平板上,筛选不能在G418抗生素平板生长而能在不含G418抗生素平板上生长的酵母菌进行PCR 验证,并传代培养15 次以上,再次用200 μg/mL zeocin抗生素平板挑选pSH65质粒丢失、无抗性标记的重组酵母菌XFa7-B/XFα6-B和XFa7-TB/XFα6-TB。基因敲除过程如图1所示。

图1 BAT2 基因敲除过程Fig.1 BAT2 gene knockout process
1.3.4 双倍体融合
将去掉抗性标记的单倍体酵母菌株XF1a7-B/XF1α6-B和XF1a7-TB/XF1α6-TB各取100 μL接种于含有3 mL YPD 的试管中摇床震荡培养8 h,取10 μL 菌液在显微镜下观察细胞融合形态,将有哑铃型的酵母融合菌液稀释涂布于YPD 平板30 ℃倒置培养2 d,挑取单菌落显微镜观察,并分散椭圆菌落进行产孢培养验证双倍体XF1-B 和XF1-TB,用引物MAT-F/MAT-α/MAT-a[18]进行菌落PCR 验证。
1.3.5 黄酒发酵
为减少干扰,本实验采用液态发酵黄酒。将浸泡后的糯米加1.6 倍体积的水蒸熟,打浆后加0.1%~0.3%耐高温淀粉酶95 ℃酶解20 min 进行液化,再加入糯米质量的5wt%麦芽、5wt%花生饼粉、250wt%水、0.25wt%中性蛋白酶、0.25%风味蛋白酶和0.1%~0.3%(V/V)糖化酶进行糖化,条件为:50 ℃糖化4 h、65℃ 1 h 和72 ℃ 20 min。过滤灭菌冷却后备用。将酵母菌接种到10 mL 上述制备的黄酒发酵液中,30 ℃摇床培养24 h 后,取1 mL 菌液接种到含50 mL 黄酒发酵液中,30 ℃摇床培养16 h,得到种子液。取100 mL黄酒发酵液接种3%(V/V)酵母菌种子液30 ℃发酵7 d。
1.3.6 生长曲线
生长曲线参照徐佳等[10]的方法,取斜面菌种1 环,接到10 mL YPD 液体培养基中,30 ℃,180 r/min 培养12 h。再以1%接种量转接至50 mL YPD 液体培养基继续培养,每隔2 h 测定600 nm 处的吸光值。
1.3.7 理化性质测定
CO2释放量采用称重法测量[18],总酸、总糖、氨基态氮的测定参照国标GB/T 13662-2018[19],酒精度的测定参照孟庆顺等[20]的方法。
1.3.8 高级醇和氨基酸的测定
高级醇的测定参照Wu 等[21]的方法通过气相色谱内标法测定。氨基酸的测定参照芮鸿飞等[22]的方法采用液相色谱测定。
1.4 数据处理
所有实验均至少重复3 次,结果以平均数±标准差表示,并通过SPSS 25.0 软件采用单因素方差分析法(ANOVA)进行显著性分析。其中,柱状图、生长曲线使用Graph Pad Prism 8.0 软件进行分析作图。
2 结果与讨论
2.1 敲除组件和重组酵母菌的构建与验证
根据1.3.1 的方法构建敲除组件,以提取的酵母基因组DNA 为模板,用引物BAT2-F1/BAT2-R1 和BAT2-F2/BAT2-R2分别PCR扩增出同源臂BU和BD;以pUG6 质粒为模板,用引物loxP-F/loxP-R 扩增出loxp-KanMX-loxp,结果如图2a、2b 所示,左同源臂BU(290 bp)的和右同源臂BD(361 bp)和抗性标记loxP-KanMX-loxP(1 736 bp)通过融和PCR 连接,得到BAT2基因敲除组件BU-loxp-KanMX-loxp-BD(2 338 bp)。


图2 敲除组件和重组菌株的PCR 验证Fig.2 PCR verification of disruption cassette
根据 1.3.2 的方法通过同源重组转化酵母XF1a7/XF1α6 和XF1a7-T/XF1α6-T,然后挑选阳性菌落进行PCR 验证,结果如图2c 所示(以XF1a7-TBK、XF1α6-TBK 示例),重组菌株2 600 bp,原始菌株为1 692 bp,PCR 验证条带和理论值一致,成功获得BAT2和THI3/BAT2基因缺失重组酵母菌。
2.2 KanMX 抗性筛选标记与pSH65 质粒丢失验证
根据1.3.3 的方法对重组菌诱导培养,然后对去除抗性的重组菌的进行PCR 验证,验证结果如图3a 所示(以XF1a7-TB 为例)。以BAT2-A 和BAT2-D 作为引物,未去除抗性的XF1a7-TBK 的PCR 产物大小为2 600 bp、去除抗性的XF1a7-TB 的PCR 产物大小为1 093 bp。以BAT2-A 和B-M 以及M-B 和BAT2-D 为引物的XF1a7-TB 的PCR 结果为无条带。结果表明重组菌的KanMX抗性基因成功去除。


图3 重组菌株XF1a7-TB 的抗性去除验证与质粒丢失验证Fig.3 Resistant removal verification and plasmid loss verification of strains XF1a7-TB
根据1.3.3 的方法将传代去除 pSH65 质粒的重组菌进行影印平板法筛选去除质粒的菌株。质粒丢失的菌株只能在不含抗生素平板上生长,不能在含zeocin抗生素的平板上生长。结果如图3b 所示(以XF1a7-TB为例),不含zeocin 抗生素的平板上重组菌生长,含zeocin 抗生素的平板上重组菌未生长。结果表明pSH65 质粒成功丢失。
2.3 基因缺失双倍体融合的重组酵母菌
根据 1.3.4 的方法将不同配型单倍体XF1a7-B/XF1α6-B 和XF1a7-TB/XF1α6-TB 融合后,以引物MAT-F/MAT-α/MAT-a 进行菌落PCR 验证,结果如图4所示,泳道1 和泳道3 中的XF1a7-B 和XF1a7-TB 均为544 bp,泳道2 和泳道4 中的XF1α6-B和XF1α6-TB 均为404 bp,泳道5 和泳道6 均有544 bp和404 bp 两条条带,表明双倍体XF1-B 和XF1-TB 融合成功。

图4 融合双倍体菌株验证Fig.4 PCR verification of Diploid strains
2.4 基因敲除对重组菌生长性能的影响
根据1.3.6 的方法绘制原始菌XF1、重组菌XF1-B和XF1-TB 生长曲线,结果如图5所示,双基因缺失酵母XF1-B 和XF1-TB 的前期的生长速率略低于XF1,但是最终生物量都没有明显差异。THI3和BAT2基因缺失影响酵母对营养物质的吸收,使其生长速率变慢,但对酵母菌体生长性能影响不大。

图5 原始菌XF1、重组菌XF1-B 和XF1-TB 生长曲线Fig.5 The growth curves of XF1,XF1-B and XF1-TB
2.5 基因敲除对重组菌发酵性能的影响
根据1.3.7 的方法测定原始菌XF1、重组菌XF1-B和XF1-TB 基本发酵性能如表3所示。从表3可知,重组菌XF1-B 和XF1-TB 的CO2产生量、总糖、总酸和酒精度与原始菌XF1 相比都无差异,说明THI3和BAT2基因缺失不影响酵母的基本发酵性能。而XF1-B 和XF1-TB 的氨基态氮分别为0.66 g/L 和0.65 g/L,提高了11.86%和10.17%,说明敲除BAT2能明显抑制支链氨基酸分解合成高级醇途径,从而使得氨基态氮积累。
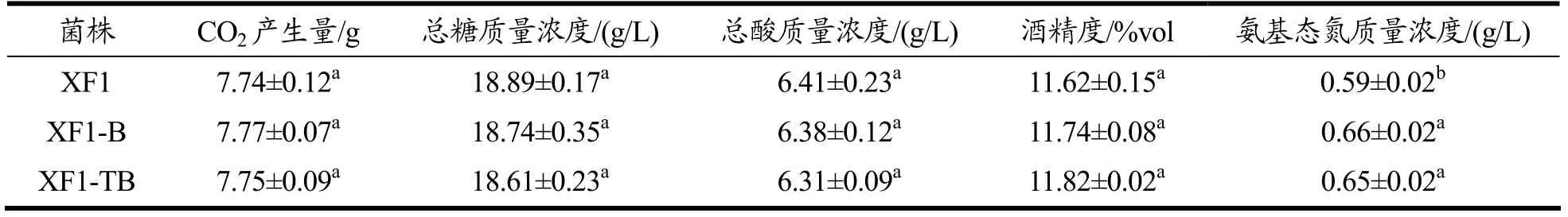
表3 原始菌XF1、重组菌XF1-B 和XF1-TB 基本发酵性能Table 3 Basic fermentation performance of XF1,XF1-B and XF1-TB
2.6 基因敲除对重组菌生成高级醇的影响
根据1.3.7 的方法测定原始菌XF1、重组菌XF1-B和XF1-TB 发酵黄酒后的高级醇和氨基酸质量浓度如图6所示。从图6可知,重组菌XF1-B、XF1-TB 发酵黄酒生成的异戊醇分别为139.60、126.06 mg/L,与原始菌XF1 相比分别降低了18.12%、26.06%;异丁醇分别为43.02、53.90 mg/L,降低了35.21%、18.83%;正丙醇分别为37.70、43.36 mg/L,提高了15.42%和32.75%;总高级醇分别为269.59、270.77 mg/L,降低了15.65%和15.28%;缬氨酸分别为286.89、281.63 mg/L,提高了6.75%、4.80%;苏氨酸分别为112.24、107.49 mg/L,提高了2.28%、6.14%;亮氨酸分别为255.98、260.71 mg/L,提高了6.97%、8.95%;异戊醇/异丁醇分别为3.24 和2.34。可见,重组菌XF1-B、XF1-TB 酿造黄酒中总高级醇降低效果明显,但是异戊醇/异丁醇比值相差大。

图6 原始菌XF1、重组菌XF1-B 和XF1-TB 发酵黄酒后的高级醇和氨基酸含量Fig.6 The higher alcohol and amino acid content in fermented Huangjiu of strains XF1,XF1-B and XF1-TB
2.7 讨论
本研究构建重组菌XF1-B 使发酵黄酒中异戊醇、异丁醇分别降低了18.12%、35.21%,与Zhang 等[23]敲除酿酒酵母BAT2基因分别降低了黄酒中异戊醇与异丁醇14.20%、33.00%的结果相似,与葛峻伶等[24]敲除啤酒酵母BAT2基因分别降低了啤酒中异戊醇与异丁醇9.55%、10.63%的结果相差较大,可能是因为发酵原料以及酵母种类不同。对应酿造黄酒中缬氨酸和亮氨酸提高了6.75%、6.97%,根据高级醇代谢途径图7分析,证实BAT2基因编码的支链氨基酸转移酶对支链氨基酸转化为高级醇途径起主要作用。
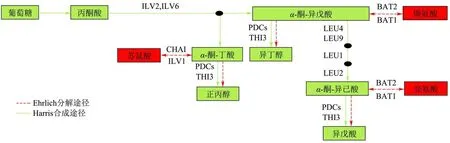
图7 高级醇代谢途径Fig.7 Higher alcohol metabolic pathway
本研究构建重组菌XF1-TB 使发酵黄酒中异戊醇、异丁醇分别降低了26.06%、18.83%。Dickinson等[25]敲除酿酒酵母THI3基因接种以亮氨酸为唯一氮源的培养基进行发酵,异戊醇降低94%,接种以缬氨酸为唯一氮源的培养基进行发酵,异丁醇由3 020 mg/L 上升为3 080 mg/L[26]。而郝欣等[27]敲除THI3基因后接种玉米汁发酵,异戊醇并无明显变化,并猜测该合成途径受到破坏,异戊醇的合成会转向氨基酸转氨途径,本研究证实在THI3和BAT2同时缺失会导致异戊醇进一步减少,并与Styger 等[28]敲除酿酒酵母THI3与BAT2双基因异戊醇与异丁醇都有降低结论一致。重组菌BAT2/THI3基因缺失时,因THI3编码的类酮酸脱羧酶是亮氨酸分解代谢途径中α-酮异己酸脱羧生成异戊醇的关键酶[12],该途径受阻导致异戊醇生成量显著降低。重组菌XF1-TB 与XF1-B 相比异丁醇提高了25.29%,目前还未见BAT2/THI3双基因缺失较BAT2单基因缺失异丁醇会提高的报道,对于α-酮异戊酸生成异丁醇的途径,因3 种同工酶(Pdc1p、Pdc5p或Pdc6p)中任何一种酶都能使α-酮异戊酸脱羧[27]而受影响小,异戊醇合成途径受阻导致α-酮酸积累,而氨基酸的增加抑制了α-酮酸合成氨基酸,使积累的α-酮异戊酸在同工酶的作用下向异丁醇途径合成,异丁醇生成量显著提高,则重组菌XF1-TB 酿造黄酒中异戊醇/异丁醇比值降低。黄酒中高级醇含量并不是越低越好,它是黄酒醇厚感的重要成分,黄酒中高级醇含量在80~540 mg/L 之间比较合适[7],异戊醇/异丁醇比值在0.5~2.5 之间醉度较低[29],适当的高级醇含量和不同高级醇比例是影响黄酒品质的重要因素。通过构建重组菌XF1-TB 酿造黄酒的总高级醇、异戊醇/异丁醇比值都显著降低,有助于降低黄酒的“上头”醉度,提升黄酒品质。
3 结论
本研究成功构建BAT2基因缺失重组酵母菌XF1-B和BAT2/THI3双基因缺失重组酵母菌XF1-TB。结果表明,重组菌XF1-B、XF1-TB 都能有效降低发酵黄酒中总高级醇含量,分别降低了15.65%、15.28%,同时对应提高黄酒中氨基氮含量约11.86%、10.17%。这表明降低发酵黄酒中高级醇含量和提高黄酒中氨基酸等营养物质含量在技术上是可行的。重组菌XF1-TB 比重组菌XF1-B 优化了不同高级醇含量,即异戊醇/异丁醇比值由3.24 降低为2.34,这进一步降低了黄酒“上头”的醉度,为提高黄酒品质提供了新的技术思路。因此,可通过构建多基因缺失的重组酵母菌来降低酿造黄酒中高级醇含量和优化不同高级醇比例,从而有效降低黄酒醉度和提高黄酒品质。
