4种藤茶多糖的检测方法比较
2022-12-14黄耀宗郭清泉焦文娟赵甜甜张友胜黄国滋
黄耀宗,郭清泉,焦文娟,赵甜甜,张友胜*,黄国滋
(1.广东工业大学轻工化工学院,广东广州 511400)(2.广东省农业科学院蚕业与农产品加工研究所,农业部功能食品重点实验室,广东省农产品加工重点实验室,广东广州 510610)(3.广东省农业科学院茶叶研究所,广东广州 510640)
藤茶,又称莓茶、张家界莓茶、连州白茶、长寿藤,是一种药食同源的藤本植物,学名为显齿蛇葡萄植物(Ampelopsis grossedentata),广泛分布于中国南部山区及平原地带。在《饮膳正要》(1330)、《草木便方》(1873)、《广西植物名录》(1971)等历代植物志中及药用植物名录中均有记载[1]。藤茶具有止咳、化痰、解痉消炎、预防上呼吸道感染、抗氧化和清除自由基、防止肝功能退化、预防酒精性肝损伤、增强免疫力等作用,是一种具有营养、保健、药用功能的类茶植物[2-5]。藤茶既含有丰富的蛋白质(11%~15%)、17 种游离氨基酸、17 种矿物质以及胡萝卜素(5.23 mg/100 g)、维生素E(6.32 mg/100 g)等多种营养成分,又含有黄酮和多糖两大类活性成分。藤茶中的总黄酮质量分数一般为35%~45%,其中单体黄酮类化合物二氢杨梅素一般为25%~35%,在目前发现的所有植物中,藤茶所具有的黄酮含量最高,被誉为“黄酮之王”[6-9]。
藤茶多糖是藤茶中另一主要活性成分。随着研究的不断深入,逐渐发现藤茶多糖具有如免疫调节功能、降低致病菌的繁殖速度、抗感染、降血糖、降血脂等多种的生物活性功能[10-13]。
从2013年藤茶被批准为新资源食品,2015年把“藤茶提取物”收录进化妆品原料目录中,藤茶多糖被许多企业作为原料进行深加工研发产品。与其它植物多糖一样,藤茶多糖不是一种单一的化学物质,而是由各种单糖、糖醛酸和蛋白质等不同的物质聚合一起形成的复合物。罗祖友等[14]对藤茶多糖中的两个纯化多糖组分进行了结构鉴定,结果表明两种藤茶多糖组分均含有中性糖(57.6%和46.2%)、糖醛酸(32.3%和45.7%),蛋白质(3.5%和4.7%)。由于多糖本身不具备还原性和变旋现象,其含量需要经过一定的处理后才能检测得出。目前比色法、滴定法、色谱法、高效毛细管电泳法等都是目前较为常用的多糖含量检测方法[15,16],但时至今日,许多文献报道的各种方法由于单糖留存较多,干扰较大,常常使结果出现较大的误差,不同的多糖所适用的检测方法也不同,所以尚无统一的多糖测定的国家标准方法。鉴于此,本研究选择目前常用的苯酚-硫酸法、蒽酮-硫酸法、硫酸-紫外法和二硝基水杨酸(DNS)法等4 种多糖常用检测方法,通过光谱扫描,单因素试验、方法学考察,比较分析四种方法测定结果的准确性和稳定性,旨在确定出最佳的藤茶多糖测定方法。
1 材料与方法
1.1 原料与试剂
藤茶原茶,由湖南乾坤生物科技有限公司提供;葡萄糖标准品,上海阿拉丁生化科技股份有限公司;浓硫酸、苯酚、蒽酮、DNS(3,5-二硝基水杨酸),天津科密欧化学试剂有限公司;其它所用试剂均为国产分析纯。
1.2 仪器与设备
UV-1800 型紫外分光光度计,日本岛津有限公司;RE-52AA 型旋转蒸发器,上海亚荣生化仪器厂;SHZ-D(Ⅲ)循环水式真空泵,巩义市予华仪器有限责任公司;Milipore 超纯水净化系统,美国Milipore超纯水系统。
1.3 藤茶粗多糖的提取及粗多糖溶液的制备
称取200 g 藤茶原茶,以1:6 的料液比加入超纯水,在100 ℃水浴中提取1 h,过滤,重复2 次,合并滤液;将滤液置于4 ℃冰箱中过夜,在4 000 r/min条件下离心20 min 去沉淀;取上清液旋转浓缩至原体积的1/2 后重复4 ℃冰箱静置过夜,在4 000 r/min 条件下离心20 min 后重新去沉淀。取上清液后加入三倍体积无水乙醇,室温放置24 h,在4 000 r/min 条件下离心20 min,收集沉淀,冷冻干燥得藤茶粗多糖[7,17]。
取500.0 mg 上述干燥所得藤茶粗多糖加入1 000 mL 蒸馏水中,充分摇匀搅拌溶解,即得浓度为0.5 mg/mL 的藤茶粗多糖溶液。
1.4 相关标准溶液的配制
1.4.1 葡萄糖标准溶液的配制
称取200 mg 葡萄糖标准品,用超纯水定容至20.0 mL,配得10.0 mg/mL 标准溶液,放入4 ℃冰箱备用。分别量取0.5、1.0、1.5、2.0 和2.5 mL 标准溶液置于50 mL 容量瓶中用水稀释至刻度线,摇匀,即得浓度0.1、0.2、0.3、0.4 mg/mL 和0.5 mg/mL 的葡萄糖标准溶液。
1.4.2 苯酚溶液的配制
准确称取5.0 g 苯酚,用体积分数30%乙醇定容于100 mL 棕色的容量瓶中,混匀即得5wt%苯酚溶液(现配现用)。
1.4.3 蒽酮-硫酸溶液的配制
准确称取蒽酮50.0 mg置于100 mL棕色的容量瓶中,浓硫酸定容,混合即得0.5 mg/mL 的蒽酮-硫酸溶液(现用现配)。
1.4.4 DNS 试剂的配制
准确称取DNS 3.15 g,加入500 mL 超纯水,水浴至45 ℃后逐步加入100 mL 0.2 g/mL的氢氧化钠溶液,不断搅拌直至溶液清澈透明。再加入四水酒石酸钾钠91.0 g、苯酚2.50 g 和无水硫酸钠2.50 g,再加入300 mL 超纯水,45 ℃水浴搅拌至完全溶解,室温冷却用超纯水定容至1 000 mL,混匀后贮于棕色瓶中避光保存7 d。
1.5 四种检测方法的比较测定
1.5.1 苯酚-硫酸法
用移液枪分别吸取“1.3”项下制备的0.5 mL 藤茶粗多糖溶液和“1.4.1”项下制备的0.5 mg/mL 的葡萄糖标准溶液于2.0 mL 离心管中,室温下分别加入50 mg/mL 苯酚溶液200~1 000 μL 摇匀,缓慢加入浓硫酸300~700 μL,混合后加水定容至2.0 mL,置于50~90 ℃下水浴显色5~40 min,冷水浴冷却,在485.5 nm 下分别测定吸光值,以获得最佳的苯酚加入量、浓硫酸加入量、水浴温度和水浴时间等实验参数。然后通过吸取不同浓度的葡萄糖标准溶液,按上述最佳实验参数进行操作,在485.5 nm 下测定吸光度,绘制苯酚-硫酸法中使用的标准曲线,按标准曲线最后测定藤茶粗多糖溶液吸光度并计算其多糖含量[18]。
1.5.2 蒽酮-硫酸法
用移液枪分别吸取“1.3”项下制备的0.5 mL 藤茶粗多糖溶液和“1.4.1”项下制备的0.5 mg/mL 的葡萄糖标准溶液于2.0 mL 离心管中,冰水浴中加入1.4.3 制备的0.5 mg/mL 蒽酮-硫酸溶液200~600 μL,摇匀,置于60~100 ℃下水浴显色5~40 min 后迅速置于冰水中冷却,在620 nm 下分别测定吸光值,以获得最佳的蒽酮-硫酸溶液加入量、水浴温度和水浴时间等实验参数。然后通过吸取不同浓度的葡萄糖标准溶液,按上述最佳实验参数进行操作,在620 nm 下测定吸光度,绘制蒽酮-硫酸法中使用的标准曲线,按标准曲线最后测定藤茶粗多糖溶液吸光度并计算其多糖含量[18]。
1.5.3 硫酸-紫外法
用移液枪分别吸取“1.3”项下制备的0.5 mL 藤茶粗多糖溶液和“1.4.1”项下制备的0.5 mg/mL 的葡萄糖标准溶液于2.0 mL 离心管中,快速加入浓硫酸300~700 μL,置于25~60 ℃下水浴5~40 min,摇匀30 s 后放入冰水沙浴中快速冷却,加水至2.0 mL,在315 nm 下分别测定吸光值,以获得最佳的浓硫酸加入量、水浴温度和水浴时间等实验参数。然后通过吸取不同浓度的葡萄糖标准溶液,按上述最佳实验参数进行操作,在315 nm 下测定吸光度,绘制硫酸-紫外法中使用的标准曲线,按标准曲线最后测定藤茶粗多糖溶液吸光度并计算其多糖含量[19]。
1.5.4 DNS 法
用移液枪分别吸取“1.3”项下制备的0.5 mL 藤茶粗多糖溶液和“1.4.1”项下制备的0.5 mg/mL 的葡萄糖标准溶液于2.0 mL 离心管中,加入DNS 溶液220~300 μL,混匀,加水至2.0 mL,置于60~100 ℃下水浴显色3~7 min 后取出,立即用冰水浴快速冷却至室温,摇匀后在510 nm 下分别测定吸光值,以获得最佳的DNS 溶液加入量、水浴温度和水浴时间等实验参数。然后通过吸取不同浓度的葡萄糖标准溶液,按上述最佳实验参数进行操作,在510 nm 下测定吸光度,绘制标准曲线,按标准曲线最后测定藤茶粗多糖溶液吸光度并计算其多糖含量[20]。
1.6 方法学考察
1.6.1 精密度测定
取0.5 mL 藤茶粗多糖样品溶液,分别用“1.5”项四种方法的最佳参数处理后在最大吸收波长处测定吸光度,同一样品溶液在2 min 内连续测定6 次吸光度,计算结果的RSD 值。
1.6.2 重复性测定
称取1.0 g 藤茶粗多糖5 份,制备成样品溶液后分别用“1.5”项四种方法的最佳参数处理后在最大吸收波长处测定吸光度,计算结果的RSD 值。
1.6.3 加标回收率测定
取5 份已知含量的藤茶多糖样品溶液,加入一定量的葡萄糖标准溶液后分别用“1.5”项四种方法的最佳参数处理后在最大吸收波长处测定吸光度,计算结果的RSD 值。
1.6.4 稳定性测定
取0.5 mL 藤茶粗多糖样品溶液,分别用“1.3”项四种方法的最佳参数处理后在最大吸收波长处每隔20 min 测定吸光度,共测定6 次(120 min),计算结果的RSD 值。
1.7 不同藤茶样品中多糖含量的测定
按“1.3”项下的方法进行样品多糖的提取,按“1.5”项下所获得最佳参数方法和步骤进行多糖含量测定。
1.8 数据处理与结果分析
所有分析测试结果均采用三次平行处理;采用SPSS 26.0 软件包进行数据处理;采用Origin 9 软件进行绘图处理;显著性水平取0.05,数据表示形式为平均值±标准差。
2 结果与讨论
2.1 四种方法最适吸收波长的确定
用四种检测方法分别处理葡萄糖溶液和藤茶多糖溶液后,用紫外光谱检测220~700 nm 波长内的吸光度。由扫描结果(图1)可知,苯酚-硫酸法、蒽酮-硫酸法、硫酸-紫外法和DNS 法的最适吸收波长分别为485.5、620、315 和510 nm。与文献报道结果相似[18-20]。另外,从出峰波长及对称性来看,苯酚-硫酸法、(图1a)、二硝基水杨酸(DNS)法(图1d)可能比其它2 种更适合测定藤茶样品中总多糖的含量。


图1 四种方法在220~700 nm 波长处的紫外扫描图Fig.1 The results of ultraviolet scanning of four methods at 220~700 nm
2.2 四种方法最佳实验条件的确定
2.2.1 苯酚-硫酸法
苯酚-硫酸法是多糖在硫酸的作用下生成糖醛衍生物,然后与苯酚生成橙黄色化合物,在485.5 nm 处有一最大紫外吸收峰,随着多糖含量的增加,其颜色和吸光度也会递增。由此可见,苯酚-硫酸法测定的多糖结果是否准确、稳定与实验过程中加入的硫酸数量、苯酚数量、生成橙黄色化合物时的反应温度和反应时间等4 个关键因素具有密切关系[21]。
由图2可以看出苯酚-硫酸法中4个关键检测条件的单因素实验对吸光值的影响结果。在0.5 mL 藤茶粗多糖溶液(浓度为0.5 mg/mL)体系中,随着浓硫酸添加量逐渐增加,与各添加量对应的吸光值显示出增加后持平的趋势,当浓硫酸的添加量为500 μL 时吸光时最大,进一步增加时的吸光值没有发现显著的差异(p<0.05);逐步增加50 mg/mL 苯酚溶液的加入量,各加入量对应的吸光值呈现先增加后缓慢减少的趋势,在加入量为600 μL 时吸光值为最大,数据之间具有显著性差异(p<0.05);当水浴时间达到20 min 时,对应吸光值最大,即使延长水浴时间,吸光值也没有显著差异(p<0.05);随着水浴温度变高,与各温度对应吸光值有增加的趋势,温度达到60 ℃时达到最大吸光值,之后的吸光值没有显著差异(p<0.05)。因此,利用苯酚-硫酸法测定藤茶粗多糖含量,综合考虑吸光值大小、实验时间和操作难易程度,其最佳实验条件为500 μL 浓硫酸、600 μL 苯酚溶液、60 ℃水浴温度和20 min 水浴时间。


图2 苯酚-硫酸法中检测条件对吸光值的影响Fig.2 Influence of detection conditions on absorbance value in phenol-sulfuric acid method
本实验结果与杨勤等[22]用苯酚-硫酸法测定地参多糖、谢建华等[23]用苯酚-硫酸法测定青钱柳多糖的研究相比,反应温度和反应时间存在差异,其原因可能在于一是反应体系内多糖种类、含量之间存在差异;二是部分实验没有对测定的吸光值进行差异性分析,事实上尽管数值的绝对值仍在变化,但数值之间并没有显著性差异。尤其就苯酚-硫酸法而言,反应为放热反应,反应过程中产生的热量能够保证反应的正常进行,过高的温度不利于操作,选择适当低温并不影响实验结果。
2.2.2 蒽酮-硫酸法
蒽酮-硫酸法其原理与苯酚-硫酸法类似,多糖在硫酸的作用下生成糖醛衍生物(羟甲基糖醛),再与蒽酮脱水缩合成蓝绿色化合物,通过紫外扫描发现该物质在620 nm 处有最大吸收峰,其颜色的深浅和吸光值与多糖含量呈正比。由原理可知蒽酮-硫酸试剂的加入量、水浴温度和水浴时间是影响实验结果的关键条件[24]。
由图3可以看出蒽酮-硫酸法中3 个关键检测条件的单因素实验对吸光值的影响结果。在0.5 mL 藤茶粗多糖溶液(浓度为0.5 mg/mL)体系中,逐步增加0.5 mg/mL 的蒽酮-硫酸溶液的加入量,各加入量对应的吸光值呈现先增加后平稳的趋势,当加入量达到400 μL 时对应的吸光值最大,增大加入量,吸光值无显著性差异(p<0.05);在60~100 ℃水浴温度范围内,各温度对应的吸光值均有显著性差异(p<0.05)且温度越高吸光值越大,说明水浴温度对吸光值的影响很大,实验要求在沸水状态下进行;当水浴时间达到20 min 时,吸光值最大,延长水浴时间,对应的吸光值呈缓慢下降趋势。因此,利用蒽酮-硫酸法测定藤茶粗多糖含量,综合考虑吸光值大小、实验时间和试剂用量,其最佳实验条件为0.5 mg/mL 蒽酮-硫酸溶液400 μL、100 ℃水浴、水浴时间20 min。

图3 蒽酮-硫酸法中检测条件对吸光值的影响Fig.3 Influence of detection conditions on absorbance value in anthrone-sulfuric acid method
本实验结果与张红等[25]利用蒽酮-硫酸法测定桑叶中多糖的含量、王文洁等[26]利用蒽酮-硫酸法测定凉粉草多糖所获得的蒽酮-硫酸用量和反应温度两个条件有所不同。其主要原因在于多糖具有多种反应体系,蒽酮-硫酸的用量取决于多糖种类及含量,所以不能一概而论;另外糖醛衍生物与蒽酮反应需要合适的温度以及合适的时间,温度高,时间可以缩短,温度低,相应反应时间要延长。
2.2.3 硫酸-紫外法
硫酸-紫外法是2013年Ammar等[27]以简化实验步骤、缩短实验时间以及避免因苯酚-硫酸法中所使用的苯酚给人体及环境带来危害为目的而改进的一种方法。硫酸-紫外法是直接利用强酸水解多糖,利用多糖水解后所产生的糖醛或其衍生物在315 nm 波长处具有紫外吸收的特点,通过绘制标准曲线对多糖进行定量测定。应用硫酸-紫外法测定多糖,实验过程中的浓硫酸用量和反应时的温度、时间是对结果产生影响的主要因素。
由图4可以看出,浓硫酸用量少于或超过500 μL时对吸光值产生明显影响,其原因可能在于浓硫酸用量过多时,过量的浓硫酸可能对315 nm 波长处有紫外吸收的产物进行继续水解形成没有紫外吸收的产物;而浓硫酸用量过少时,部分多糖可能没有得到水解。在25~60 ℃水浴温度范围内,温度对吸光值没有显著差异,其原因可能在于硫酸-紫外法为放热反应,实验产生的热量足够保证反应正常进行;当水浴时间达到20 min 时,吸光值最大,延长水浴时间,对应的吸光值没有差异。因此,利用硫酸-紫外法测藤茶多糖的最佳实验条件是500 μL 浓硫酸、水浴温度25 ℃、水浴20 min。实验结果与辛敏等利用硫酸-紫外法测定千两茶中总多糖的条件基本一致[28]。

图4 硫酸紫外法中检测条件对吸光值的影响Fig.4 Influence of detection conditions on absorbance value in sulfuric acidUV method
2.2.4 DNS 法
DNS 法是还原糖能在碱性条件下与DNS 发生显色反应,在高温条件下呈棕红色,其颜色的深浅和吸光度与还原糖含量成正比。故DNS 方法适合用在多糖水解产生的多种还原糖体系中。对于DNS 法而言,DNS 用量、反应温度和反应时间是影响吸光度的主要因素。
由图5可以看出,随着DNS 用量的增加,吸光值呈现增大趋势,当加入量达到240 μL 时,吸光值达到最大;在60~100 ℃水浴温度范围内,各温度对应的吸光值均有显著性差异(p<0.05),且温度越高吸光值越大,说明水浴温度对吸光值的影响很大,实验要求在沸水状态下进行;反应时间达到5 min 时,吸光值变化呈平稳状态。因此,利用DNS 法测定藤茶粗多糖含量,在0.5 mL 藤茶粗多糖溶液(浓度为0.5 mg/mL)反应体系中,选择DNS 用量240 μL、水浴温度100 ℃,反应时间5 min 时,吸光值最大。实验结果与任婧等[20]用DNS 法测定番茄果实总糖、欧能奉等[28]测定当归多糖的反应温度和反应时间基本相同,但选择DNS 最佳用量与本研究有较大差别,其主要原因在于实验过程中反应体系中多糖的种类和含量不同所致。

图5 DNS 法中检测条件对吸光值的影响Fig.5 Influence of detection conditions on absorbance value in dinitrosalicylic acid method
2.3 四种方法标准曲线的比较
按照“1.5”项下所获得的最佳实验参数和步骤进行操作,以葡萄糖标准溶液系列浓度C(mg/mL)为横坐标,吸光度A值为纵坐标,绘制标准曲线,分别得到四种方法相对应的线性回归方程:苯酚-硫酸法:A=1.516 8C+0.091 1,R2=0.998 9,线性范围为0.1~0.6 mg/mL;蒽酮-硫酸法:A=1.445 2C+0.169 7,R2=0.998 4,线性范围为0.1~0.6 mg/mL;硫酸-紫外法:A=0.799 9C+0.294 8,R2=0.998 8,线性范围为0.1~0.6 mg/mL;DNS法:A=0.981 2C-0.021 7,R2=0.998 6,线性范围为0.1~0.6 mg/mL。四种方法的相关系数R2表明在相应的线性范围内,四种方法的标准曲线均线性良好。
2.4 四种测定方法的方法学考察结果
方法学分析结果如表1所示,四种方法精密度的RSD 值均小于1.5%,说明方法的精密度较好;四种方法重复性的RSD 值均小于2.0%,在误差允许范围内再现性良好;在120 min 实验时间内,4 种方法的稳定性RSD 值均小于1.6%,说明方法测定所基于的产物是基本稳定的;四种方法的加标回收率结果显示,相互之间并没有显著性差异。
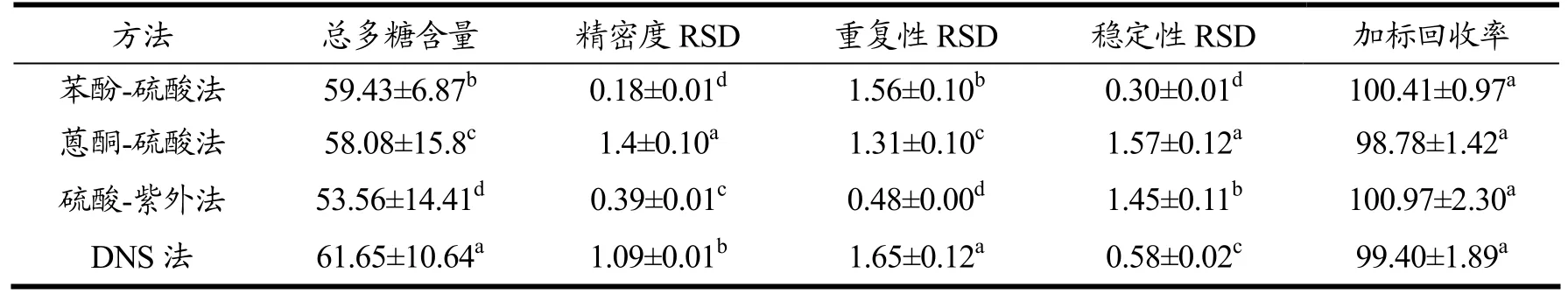
表1 四种方法测定藤茶样品中总多糖的含量及方法学考察结果Table 1 The comparison of methodological and results of polysaccharide in Ampelopsis grossedentata by four methods (%,x±SD,n=3)
综合方法学考虑结果,就藤茶多糖而言,苯酚-硫酸法是一种最好的方法,其精密度最高(RSD 为0.18%),稳定性最好(RSD 为0.30%),重复性(RSD为1.56%)、加标回收率(RSD 为100.41%)也处于4种方法的中值。其原因可能在于,苯酚-硫酸法中是分开加入试剂,试剂之间互相影响较小,加入苯酚试剂后迅速加入浓硫酸,浓硫酸与多糖生成的产物迅速与苯酚反应,浓硫酸所放出的热量足够让反应得以顺利完全进行,实验过程中的反应温度不应该是关键因素;此外,反应生成的最后产物也比较稳定,在一定时间内不会分解,所以稳定性和精密度比较好;由于反应比较彻底完全,所以重复性和回收率也比较高。
从样品总多糖含量测定的数据来看,DNS 测得的样品含量最高(61.65%),硫酸-紫外法测得样品含量最低(53.56%),苯酚-硫酸法(59.43%)和蒽酮-硫酸法(58.08%)居于中间,含量均具有显著性差异(p<0.05)。其原因可能在于DNS 法的专属性不强,显色的深浅只与多糖中还原基团的数量有关,而对还原基团的种类没有选择性,从而使得所测结果偏高[20,27];而硫酸-紫外法是直接利用强酸处理多糖,利用处理后所产生的糖醛或其衍生物在315 nm 处直接显色进行测定,但有可能部分多糖产物在315 nm 处不显色,从而使得所测结果偏低[19]。苯酚-硫酸法是利用多糖在硫酸的作用下脱水生成的糖醛衍生物与苯酚生成的橙黄色化合物,在485.5 nm 处测定吸光值,而蒽酮-硫酸法则是利用多糖在硫酸的作用下脱水生成糖醛衍生物与蒽酮脱水缩合成蓝绿色化合物,在620 nm 处测定吸光度。苯酚-硫酸法和蒽酮-硫酸法能够测定可溶性糖含量,所以测得的多糖含量居于4 种方法的中间[18,21]。
2.5 四种方法对不同样品多糖含量的测定
用苯酚硫酸法、蒽酮硫酸法、硫酸-紫外法、DNS法分别对来自相同季节、不同地点的7 批样品中的多糖含量进行了统一检测。由表2和表3可以看出,DNS法检测的多糖含量最高,硫酸-紫外法检测的多糖含量最低,7 个样品中总多糖含量幼嫩茎叶为9.87~10.47 mg/g、粗老茎叶为29.77~30.47 mg/g。从统计学来看,4 种方法检测的结果基本均存在显著性差异(p<0.05)。样品测定的多糖含量结果趋势与4 种方法的方法学考察时的结果趋势一致。
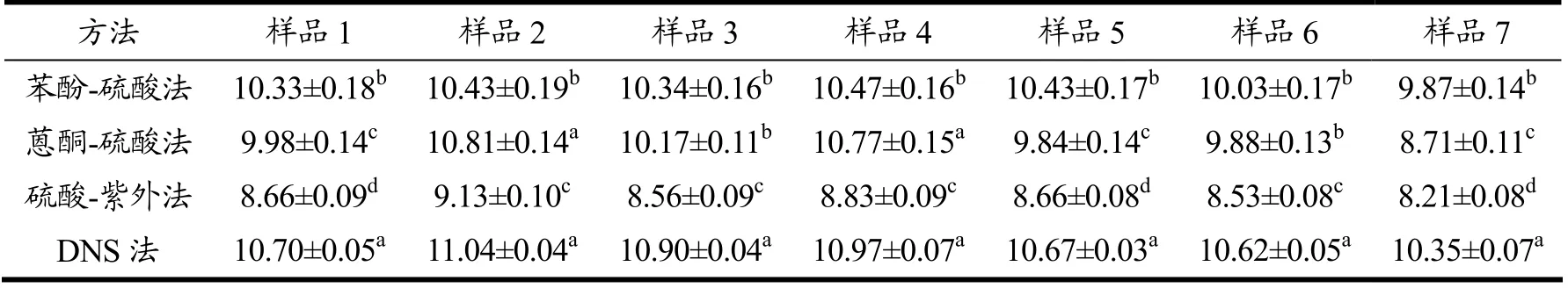
表2 不同样品中多糖含量(幼嫩茎叶,mg/g)Table 2 The polysaccharide content in different samples (young stems and leaves,mg/g)

表3 不同样品中多糖含量(粗老茎叶,mg/g)Table 3 The polysaccharide content in different samples (coarse old stems and leaves,mg/g)
3 结论
本研究以葡萄糖为对照品,选用苯酚-硫酸法、蒽酮-硫酸法、硫酸-紫外法和DNS 法对藤茶中的多糖含量进行比较测定,四种测定方法相的最大紫外吸收波长分别为485.5、620、315 和510 nm,此时样品多糖与标准品的峰形基本一致,对称性较好。四种方法的单因素试验结果表明,在0.5 mL 藤茶粗多糖溶液(浓度为0.5 mg/mL)反应体系中,苯酚-硫酸法的最佳实验条件为500 μL 浓硫酸、600 μL 苯酚溶液、60 ℃水浴温度和20 min 水浴时间,蒽酮-硫酸法的最佳实验条件为0.5 mg/mL 蒽酮-硫酸溶液400 μL、100 ℃水浴、水浴时间20 min,硫酸-紫外法的最佳实验条件为500 μL 浓硫酸、水浴温度25 ℃、水浴时间20 min,DNS 法的最佳实验条件为DNS 用量240 μL、水浴温度100 ℃,水浴时间5 min,此时吸光值最大,实验所用试剂和实验时间最少。四种方法的方法学分析结果表明,四种方法均可用于藤茶多糖含量的测定。其精密度的RSD 值均小于1.5%,方法的精密度较好;实验重复性的RSD 值均小于2.0%,在误差允许范围内,四种方法均有良好的重现性;在120 min 实验时间内,4 种方法的稳定性RSD 值均小于1.6%,说明方法测定所基于的产物是基本稳定,使用时只要严格控制测定时间方法是稳定的;加标回收率结果显示,四种方法之间数据没有显著性差异。但就藤茶多糖而言,苯酚-硫酸法是最好的方法,其精密度最高(RSD为0.18%),稳定性最好(RSD 为0.30%),重复性(RSD为1.56%)、加标回收率(RSD 为100.41%)和最终测得的多糖含量数值也处于4 种方法的中值。利用4 种方法对藤茶幼嫩茎叶干燥样品进行测量,7 个样品中总多糖含量幼嫩茎叶为9.87~10.47 mg/g、粗老茎叶为29.77~30.47 mg/g。
