石墨炔:一种新型二维炭材料的合成、改性与应用
2022-12-13王宇晶于灵敏士丽敏柴守宁
张 婷,王宇晶,*,于灵敏,士丽敏,柴守宁,何 炽
(1.西安工业大学 材料与化工学院,陕西省光电功能材料与器件重点实验室,陕西 西安 710021;2.西安交通大学 能源与动力工程学院,动力工程多相流国家重点实验室,陕西 西安 710049)
1 简介
近年来,低维材料备受科研人员关注,在各个领域的基础和应用研究被广泛开展[1–5]。碳基二维材料,因具有独特的分子拓扑结构和物理化学性质,已逐步成为电子[6]、磁学[7]、光学[8]、催化[9–11]等领域的“明星”材料[12–14]。炭材料可以通过改变碳原子之间成键方式获得多种同素异形体,典型代表如石墨烯[15]、碳纳米管[16]、富勒烯[17]等,其中碳原子之间通过sp2杂化成键(C=C)。此外,碳原子还有另一种sp 杂化成键方式[18,19],形成C≡C,所构建的分子具有线性结构、高度共轭和无顺反异构体等特点[20,21],因此,科学家也一直致力于开发包含sp 杂化碳原子的新型二维炭材料。
1987 年,Baughman 等[22]理论预测了由sp 和sp2杂化碳原子共同组成的二维碳质同素异形体—石墨炔的存在。然而,直至2010 年,才由中国科学家李玉良团队成功合成了均匀连续、表面光滑平整的此类二维炭材料,即γ−石墨双炔(γ−graphdiyne,简称GDY,注:本文中无特别说明时,GDY 均代表γ−石墨双炔)薄膜[23]。如图1 所示,石墨炔的结构与石墨烯相似,却又有明显的区别。石墨炔分子是由苯环通过不同数目的C≡C 相连形成的对称单元构成,调变相邻两个苯环之间的炔键数(n),可以得到石墨单炔(n=1,graphyne)、石墨二炔(n=2,graphdiyne)、石墨三炔(n=3)等[24]。石墨炔分子中不但含有苯环,还有由苯环的单边和C≡C 构成的包含12/18/24…个碳原子的大三角形碳环。
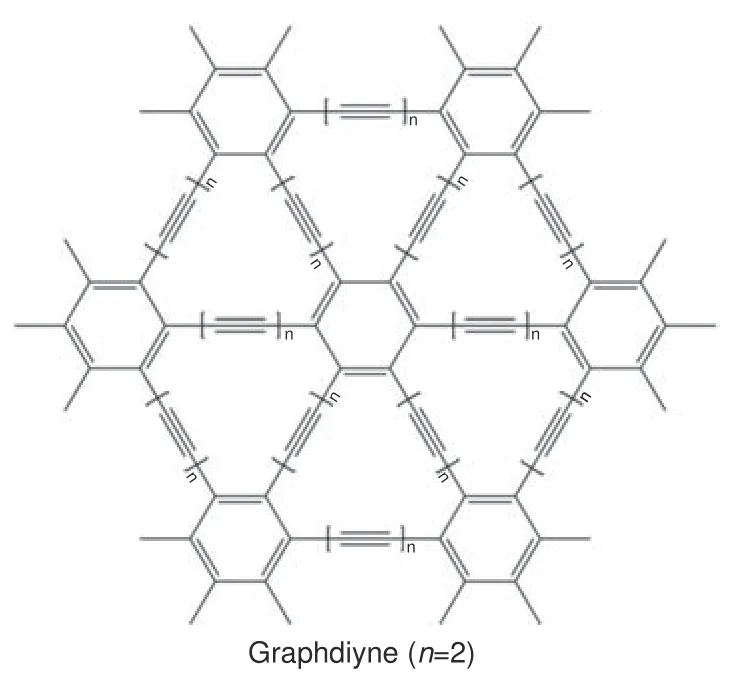
图1 GDY 的分子结构Fig.1 Molecular structure of graphdiyne
石墨炔同时拥有C≡C 和苯环结构,使其不仅具备直接带隙和狄拉克锥,还具有多重的ππ 共轭特性、规则有序的孔道和可调的电子结构等[25–27],成为继金刚石、富勒烯、碳纳米管、石墨烯之后,最具潜力的新型炭材料。近十年来,石墨炔的理论计算和实验研究均取得了显著成绩。因此,本文重点归纳石墨炔在合成、改性、应用方面的研究进展。可以预见,随着其制备方法逐渐成熟和简化,石墨炔将在众多领域展示出其独特的功用,成为继石墨烯之后的又一颗炭材料之星。
2 石墨炔的合成与表征
2.1 Glaser-Hay 反应合成法
2010 年,李玉良院士团队[23]采用湿化学法制备了大面积的GDY 薄膜,这为碳质材料的发展开辟出了新天地。具体合成路线如图2a 所示,通过铜催化六乙炔基苯(HEB)的Glaser-Hay 交叉偶联反应,成功在铜箔表面合成大面积的GDY 薄膜(3.61 cm2)。Glaser-Hay 反应是构建对称共轭二炔化合物的主流方法,机理如图2f。铜箔除了充当GDY 薄膜的生长基底外,还代替传统端炔合成常用的铜离子作为催化剂,在吡啶溶液存在的条件下,铜箔表面产生痕量Cu(II)对交叉偶联反应发挥催化作用,即端基炔在碱性条件下被夺去一个酸性C―H 质子而形成负离子,由于端基炔负离子富电子,可通过取代反应与铜配位,形成苯基炔铜配合物中间体,氧化聚合可得到偶联的二炔产物[28]。图2b-e 为铜片上制备GDY 薄膜的扫描电子显微镜(SEM)照片,可见薄膜厚度约为1 μm,均匀连续、边缘卷曲。该GDY 薄膜电导率为2.516×10−4S·m−1,在室温下表现出优异的半导体特性。
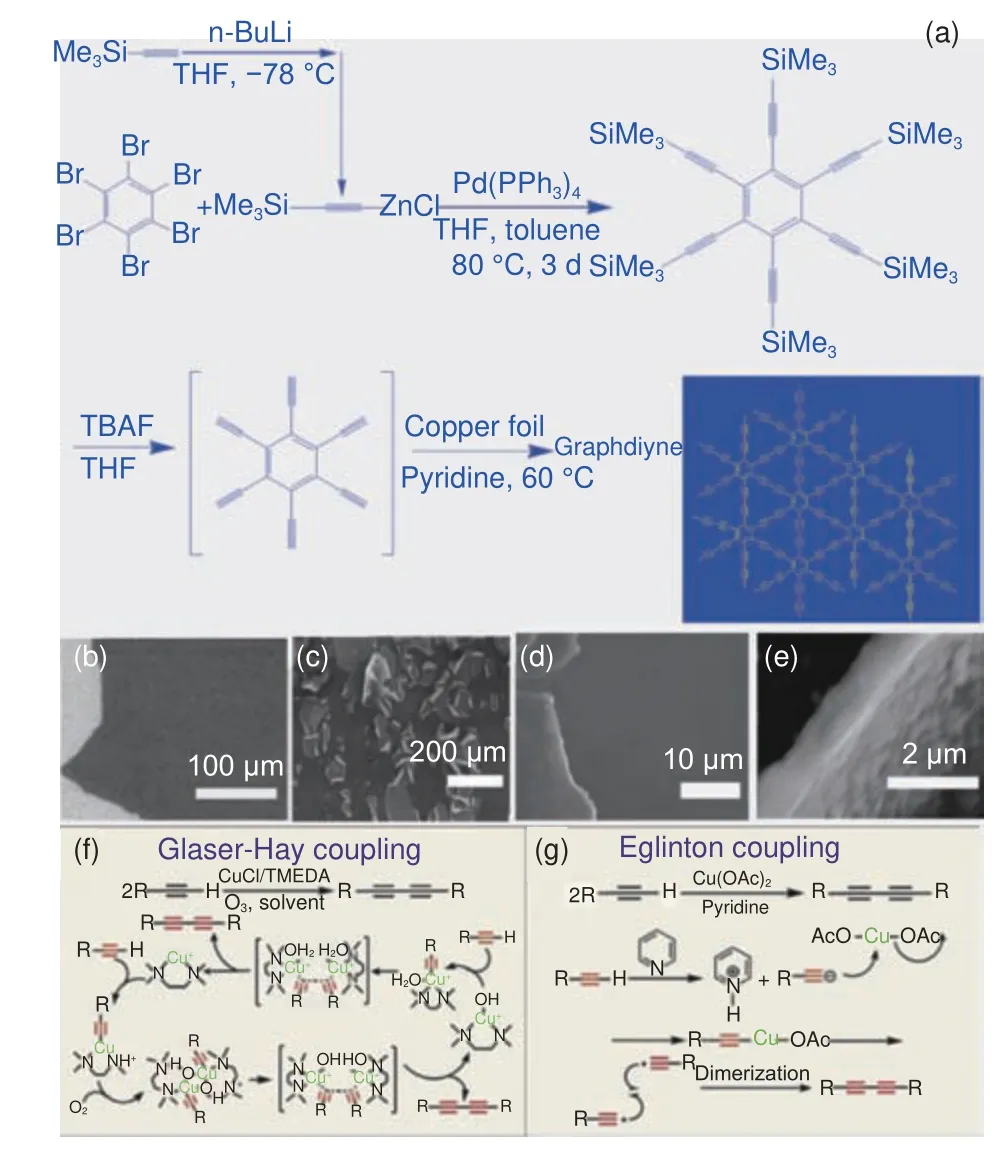
图2 (a) Glaser-Hay 交叉偶联法合成路线[23];(b-e) 石墨炔的SEM 图像[23];(f) Glaser-Hay 偶联化学反应机理;(g) Eglinton 偶联化学反应机理[28]Fig.2 (a) Synthesis route by Glaser-Hay cross coupling method [23]; (b-e)SEM images of graphdiyne[23]; Proposed mechanism for Glaser-Hay coupling reaction (f) and Eglinton coupling reaction (g)[28].Reprinted with permission
黄长水等[29]使用相同的方法,发现调变溶剂中单体的浓度可以调控GDY 薄膜的厚度。他们还研究了热处理对GDY 成膜质量和形貌的影响[30],发现表面形貌发生了从初始的粗糙粒子聚集到多孔破缺再到均匀光滑的转变。当热处理温度升至300 °C 后,GDY 薄膜中嵌入的大部分低聚物和气化温度较低的颗粒逐渐蒸发,表面形成多孔结构。随着温度继续升高,GDY 薄膜表面的小碎片发生交叉偶联反应,缺陷进行自修复,形成均匀光滑微观表面。然而,当温度高于500 °C 后,在最低表面能驱动下薄膜结构被破坏,生成GDY 球形颗粒和堆积片层。可见,热处理可以去除GDY 表面的低聚物和非晶相,实现对GDY 缺陷和瑕疵的自修复。Kong[31]等采用Cu(II)-N,N,N’,N’-四甲基乙二胺催化剂,在聚丙烯酰胺水凝胶上构建超铺展的液/液界面,以快速可控地合成GDY 薄膜。该催化剂能够高效催化末端炔基偶联,从而提高了GDY 的成核密度和生长速率。室温下2 h 内可制备出4~50 nm 厚度可控、形貌均匀的GDY 薄膜。
为满足及拓展GDY 在不同领域的应用,研究者们对Glaser-Hay 反应有机溶剂液相合成方法进行改进[32–33],成功制得不同形貌的GDY,如纳米管、纳米线、纳米棒、纳米墙等。李玉良、刘辉彪等[34]以铜箔为催化剂,电化学阳极氧化铝纳米管为模板制得GDY 纳米管阵列。Al―O 键与HEB 在氢键作用下,在氧化铝模板壁内形成一层HEB 薄膜,铜箔和吡啶形成铜-吡啶配合物催化炔基偶联,生成GDY 纳米管(GDNT)。GDNT 表面光滑,直径约为200 nm,壁厚约为40 nm。经高温退火处理后,GDNTs 的壁厚减薄到15 nm 左右。他们还以GDY 粉末为气源,ZnO 纳米棒阵列为基底和自生长催化剂,通过选择不同的GDY 粉末作为气源,调节反应温度、气源到基底的距离等,利用气-液-固(VLS)法控制合成了导电性好、电子迁移率高的GDY 纳米线[35]和GDY 纳米薄膜[36]。其机理可能为,在Ar 气氛下,GDY 气体还原ZnO 纳米棒的尖端,产生金属Zn 的小液滴。随着反应的持续,GDY 在Zn 液滴中变得过饱和,从液滴中分离出来,在Zn 液滴表面形成GDY 核。Zn 液滴的大小和数目不同及GDY 在液滴表面的浓度差异,决定着GDY 的一维生长或二维生长。近期,他们又通过原位聚合技术在氧化铝纳米管模板中合成GDY 纳米棒,然后结合原位电诱导自组装技术和电沉积技术制得核/壳结构GDY/CuS 异质结纳米棒。通过调节Cu 纳米壳的长度控制异质结界面的尺寸,而改变电化学沉积生长Cu 的时间可以很容易地调节纳米棒壳层的长度[37]。刘忠范院士团队[38]报道了一种制备GDY 纳米墙的合成方法。其形成机理如图3a 所示,通过调节有机碱和单体比例,使铜在大量碱的存在下转化为铜离子,形成催化反应位点,GDY 首先在这些位点垂直生长。图3b 揭示了GDY 纳米墙的微观生长过程。随着反应进行,反应介质中生成的铜离子越来越多,GDY 生长更加致密,最终在铜基底表面形成均匀的GDY 纳米墙。薛玉瑞等[39]在铜泡沫基底上,以铜为核,通过自催化生长GDY 壳层,制备出一种原位生长的自支撑核壳纳米线阵列(Cu@GDY NA/CF,图3c-j)。纳米带是另一种具有强烈的边缘效应和纳米效应的重要一维材料[40–42],李勇军等[43]通过乙炔基的分子内聚合和分子间偶联两步反应制备得到的宽度均匀的菱形GDY 纳米带(GDYNR,图3k-o),从AFM 可知GDYNR 的厚度约为4.5 nm,并通过π-π 堆叠作用将此纳米带交织成纳米织物。
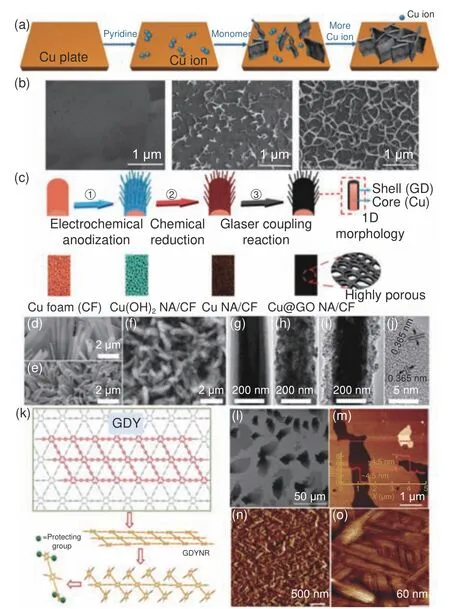
图3 (a) 石墨炔纳米壁的形成过程;(b)反应前后8、10 h 的SEM 图[38];(c) Cu@GD NA/CF 的制备示意图;(d-f) Cu(OH)2 NA/CF、Cu NA/CF、Cu@GDY NA/CF 的SEM 图像;(g-i) Cu(OH)2、Cu,、Cu@GDY 纳米线的TEM 图像;(j) Cu@GDY 纳米线的HRTEM 图像[39];(k) GDYNR 的合成策略;(l) GDYNR 的SEM 图像;(m-o) GDYNR 的AFM 图像[43]Fig.3 (a) Formation process of graphdiyne nanowall; (b) SEM images before and 8 and 10 h after the reaction[38]; (c) Schematic diagram of preparation of Cu@GD NA/CF; (d-f) SEM images of Cu(OH)2 NA/CF, Cu NA/CF and Cu@GD NA/CF; (g-h) TEM images of Cu(OH)2, Cu, and Cu@GD nanowires; (j) HRTEM image of Cu@GD nanowires[39];(k) Synthesis strategy of GDYNR; (l) SEM image of GDYNR;(m-o) AFM image of GDYNR[43].Reprinted with permission
2.2 其它合成法
除了HEB 在铜表面引发Glaser-Hay 反应合成GDY 以外,研究人员又相继开发了其它合成方法,包括化学气相沉积法、范德华外延生长法、爆炸法、界面限域合成法和双极电化学法等,如图4 所示。(1)化学气相沉积法。在液相介质中铜基底表面合成过程中,一旦铜表面被完全覆盖,常会因催化剂缺失导致反应停止,生长受限。而化学气相沉积过程中,前躯体在基体表面可以直接发生气相反应,从而避免上述问题。张锦院士团队[44]采用银箔盒作为载体,以HEB 作为前驱体,使用化学气相沉积法在银箔盒内表面成功生长单层GDY。和铜相比银表面催化副反应更少,且银的热蒸发损失较小,退火后的银箔盒内表面比外表面更光滑,有利于前驱体单体在其上的扩散。在热活化和底物催化作用下,末端的Csp―H 键的裂解使两个吸附单体之间的炔基连接形成C―C 键,最终形成规整连续的二维GDY 网络结构。(2)范德华外延生长法。上述团队[45]又开发了在石墨烯上生长超薄单晶GDY 膜的范德华外延生长策略。将石墨烯基底浸入溶解有HEB 单体的CH2Cl2介质中,HEB 分子被吸附到石墨烯上,然后滴加乙酸铜的吡啶溶液催化发生Eglinton 偶联反应(反应机理如图2g),在室温Ar 气氛下反应24 h 后,在石墨烯表面合成超薄GDY 薄膜。吡啶碱的存在促进了HEB 末端炔的脱氢和炔铜中间体的生成,通过乙酸配体桥键将单个电子转移到Cu(II)后,乙酰Cu(I)物种迅速氧化,Cu(II)配体的分解和游离炔基自由基的重组导致偶联的二炔产物生成[28]。该方法的优势在于,传统外延生长过程需要严格的晶格匹配条件,而该外延层石墨烯和吸附的HEB 反应物分子之间相互作用较弱,有效消除了GDY 和石墨烯间巨大的晶格失配的影响。然而,HEB 单体在石墨烯上的结合能却比在GDY 上更高,石墨烯上优先吸附的单体有利于面内Eglinton偶联反应。HEB 单体快速的面内耦合和缓慢的面外生长,保证了单层/准单层超薄GDY 的控制合成。(3)爆炸法。左自成等[46]在空气气氛下,不使用任何金属催化剂,对HEB 前体使用加热爆炸法成功制得GDY 粉末,产率高达98%,产品表现出优异的热稳定性和粉末导电性。该方法反应速度快、合成周期短,且避免有机溶剂及催化剂的使用。然而,在高温条件下单体不稳定,容易发生副反应,很难形成高结晶度的GDY。(4)界面限域合成法。2020 年,童廉明等[47]又提出了快速、无催化剂的微波诱导固/液界面温度梯度驱动合成法。在含有HEB 的苯/正己烷混合有机溶剂中,放入NaCl 为固体基底及吸波介质,在微波辐照作用下,NaCl 颗粒吸收能量后温度升高,而溶剂不被加热,保证了单体的稳定。NaCl固体与有机溶剂之间的固液界面处形成温度梯度,引发单体在固体表面发生交叉偶联反应,成功制得平均厚度小于2 nm 的GDY 薄膜。Nishihara 等[48]报道了GDY 的液-液和气-液界面催化限域合成。他们使用了不混溶的含有HEB 的二氯甲烷和含有催化剂(乙酸铜和吡啶)水溶液两相体系,在室温下,在HEB 和催化剂存在下,两相界面逐渐发生Eglinton 反应生成GDY 膜。GDY 薄膜具有片状外观形态,横向尺寸为25~100 mm,厚度为24 nm。类似方法,在Ar 保护下,将含有较低HEB 单体浓度的混合有机溶剂缓慢滴加到含有催化剂的水溶液表面。当有机溶剂快速挥发时,在气-水界面同样会发生偶联反应,生成GDY 纳米片漂浮在水上。(5)双极电化学法。该法是在双极电极电化学体系中进行,即在传统电解池的阴阳两极中间水平放置一块未外连的导电材料所构成的体系,与外界电源连接的阴阳极称为驱动电极,而平置的导电材料被称为双极电极[44](Bipolar electrodes, BPEs)。在驱动电极作用下,电解槽内产生均匀的电场分布,放置其中的BPEs 表面会形成电势差,从而在BPEs 两端引发氧化反应和还原反应,通过控制驱动电极可以有效地实现BPEs 表面电化学反应的调控[49–50]。Navaee 等[51]使用铜栅辅助双极电化学方法,通过施加偏压将金属Cu 氧化成Cu(I)充当催化剂,在乙醇/乙腈混合溶剂中1,3,5-三乙炔基苯单体聚合成类GDY 纳米片。
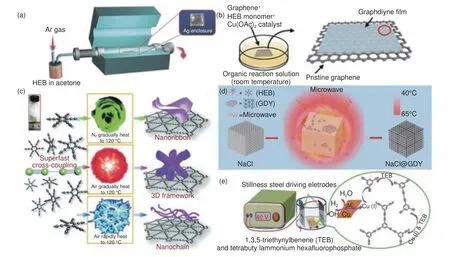
图4 (a) 化学气相沉积法合成示意图[44];(b) 液相范德华外延法合成示意图[45];(c) 爆炸法合成示意图[46];(d)界面微波诱导法合成示意图[47];(e) 双极电化学法合成示意图[51]Fig.4 Synthetic schematic diagram of (a) chemical vapor deposition method[44]; (b) liquid phase van der Waals epitaxy method[45]; (c) explosion method[46]; (d)interface microwave induction method[47]; (e) bipolar electrochemical method[51].Reprinted with permission
2.3 石墨炔表征技术
GDY 结构的精准表征和解析,不但能够指导其可控合成和改性,而且对于探究其构效关系至关重要。常见的GDY 表征技术包括三大类,第一类是用于表征微观形貌、厚度尺寸及层数,如SEM、透射电子显微镜(TEM)及原子力显微镜(AFM),如前文所述。第二类是用于表征晶体结构特征,如X 射线衍射仪(XRD)、高分辨TEM(HR-TEM)、选区电子衍射(SAED)。以使用最普遍的XRD 为例,GDY 的形貌、分子结构、结晶度等均会影响其XRD 特征峰。如GDY 二维薄膜在21.18°、23.47°、43.30°、43.44°、44.42°、50.54°等位置有明显衍射峰[23]。黄长水等合成的GDY 纳米片在26.5°处出现了唯一的宽峰[52]。而GDY 纳米带在2.1°、6.3°、10.0°、26.9°处出现特征峰[43]。利用XRD、HR-TEM 图、SAED 能够获得多层GDY 的层间距、晶格参数、堆垛方式等。第三类是用于碳碳成键形式表征,傅立叶变换红外光谱仪(FT-IR),拉曼光谱仪(Raman),X 射线光电子能谱仪(XPS),碳固体核磁仪(13CNMR),X 射线吸收谱仪(XAS)等。GDY 具有与石墨烯相似的二维平面结构,具有高π 共轭相互作用。然而,GDY 含有独特1,3-二炔键,这是石墨烯结构中所没有的。因此,依据Raman、XPS 可以对二者碳的成键形式进行区分和鉴别。GDY 的Raman 光谱有3 个主要特征峰,分别代表sp2碳原子的面内振动、层数和结构或边缘导致的缺陷。Zhang 等[53]利用理论计算猜测了GDY 的拉曼光谱形式,可以发现6 种振动模式对应的6 个峰,其中包括GDY 独特的炔键的同步和异相伸缩模式产生的两个典型峰(图5a)。图5b,c 表明,与石墨烯相似,GDY 的芳香族键在拉伸过程中出现一个峰[55],但由于富含炔烃的二维芳香族体系使其发生了红移。GDY 薄膜不同位置的拉曼光谱有4 个典型的峰。其中,在2 189.8 cm−1和1 926.2 cm−1处的峰值可以归因于共轭二炔键(―C≡C―C≡C―) 的振动。在1 382.2 cm−1处的峰是苯环中sp2杂化碳原子的呼吸振动,与石墨的D 带相比发生蓝移,是与非晶态碳和结构缺陷有关的无序带。1 569.5 cm−1处的峰对应苯环中sp2杂化碳原子的伸缩振动,与石墨的G 带(1 575 cm−1)相比发生红移,是有序带。GDY 薄膜的D/G 强度比为0.729,表明其结晶度高、低缺陷。由于GDY 结构的结晶度差和高缺陷导致分辨率差和波段更宽,虽然对GDY 的典型拉曼光谱进行了系统的探索,但至今预测光谱特征总是与实验结果并不能完全吻合。如图5d,e 所示,氧化石墨烯在XPS 的C1s 谱图上主要有4 种结合能的特征信号峰284.5、286.4、287.8 和289.0 eV,分别对应于碳碳双键和单键(C=C/C―C)、环氧基和烷氧基(C=O/C―O)、羰基(C=O)和羧基(COOH)[54]。GDY 的C1s 峰可以分为284.5、285.2、286.9 和288.5 eV 四个特征峰,分别归属为C―C (sp2)、C≡C (sp)以及由于GDY 吸附部分空气中氧气导致的C―O 和C=O。GDY 中C―C(sp2)和C≡C(sp)比例约为1∶2,说明其中苯环通过二炔键互相连接[23]。
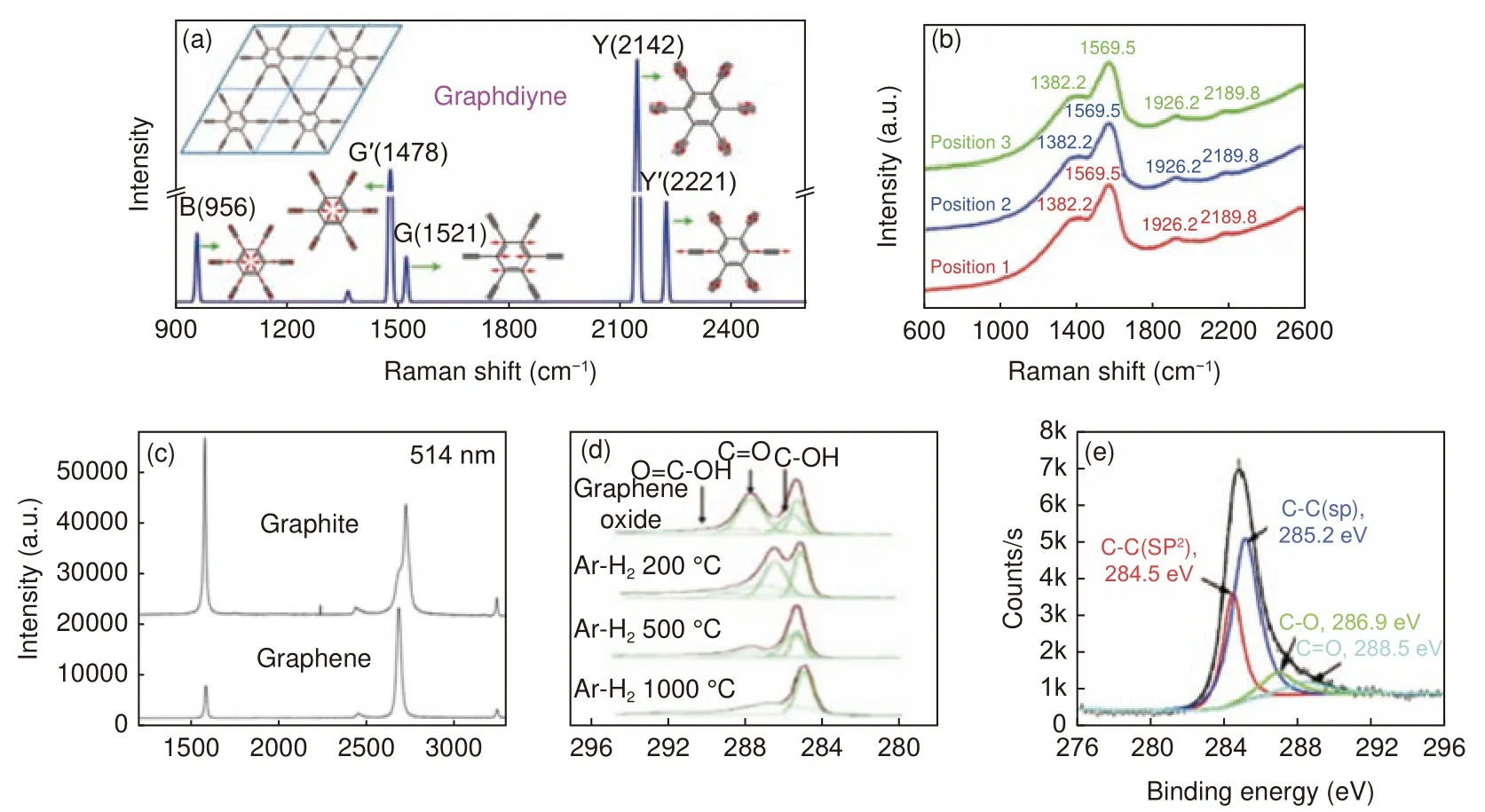
图5 (a)GDY 理论计算[53]和(b)实际样品Raman 光谱图[23];(c)石墨和石墨烯的Raman 光谱图[55];(d)氧化石墨烯[54]和(e)GDY 的XPS 谱图[23]Fig.5 (a) Calculate[53] and (b) experimental Raman spectrum of GDY[23]; (c)Experimental Raman spectra of graphite and graphene[55];XPS C1s spectra of (d) graphene oxide[54] and (e) graphdiyne[23].Reprinted with permission
3 石墨炔的改性
众所周知,对炭材料进行合理的功能化改性是其提高性能和拓展应用的重要途径。其中,最直接和简便的改性方法就是杂原子掺杂。引入杂原子不仅可以调节炭材料的电子结构,还在本体材料中引入拓扑缺陷,改变其物理化学性质。
3.1 非金属原子改性
掺杂合适的杂原子能够改善GDY 的性能,这些杂原子将通过改变能带、自旋和电荷分布直接影响GDY 的催化活性。Gu 等[56]利用理论计算系统地研究了非金属原子(B、N、P 和S)在GDY 不同掺杂位点时的氧还原反应(ORR)和析氧反应(OER)催化性能,构建的掺杂模型如图6 所示。掺杂B、N、P 或S 后,GDY 能带带隙都有不同程度的降低,这有利于催化过程中的电子转移。计算结果表明,与Pt/C 或RuO2相比,石墨型S 掺杂G D Y(S 1 型)、s p-N 掺杂G D Y(N3 型)和石墨型P 掺杂GDY(P1 型)的ORR/OER活性与其相当甚至更高,它们的ORR 催化活性高低顺序为S1 型>Pt/C>N3 型,OER 催化活性高低顺序为P1 型>RuO2>N3 型。有趣的是,N3 型掺杂GDY 可以同时拥有ORR 和OER 高活性位点,分别是N 3-C 4 原子和N 3-C 7 原子。高ORR 活性是源于其较大的正电荷(N3-C4 的净电荷约为0.06 |e|),而高OER 活性则归因于其高自旋(N3-C7 具有中等正电荷,但自旋数非常高,为0.04 μB),该研究可为掺杂型GDY 电催化剂的设计和ORR/OER 活性位点的辨识提供指导。
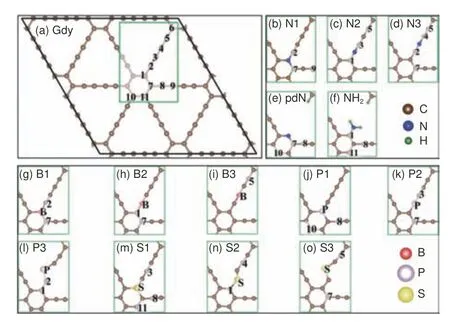
图6 原始石墨炔和不同掺杂位点N、B、P、S 掺杂石墨炔的模型[56]Fig.6 The model of original graphdiyne doped with different doping sites N, B, P, S[56].Reprinted with permission
杂原子种类、掺杂构型和掺杂量对GDY 能带带隙、电子输运性质和物化性质会产生不同的影响,研究者不但借助理论计算对GDY 的掺杂改性进行探究,而且通过基础实验对理论研究进行有效验证和补充。N 原子掺杂是改善碳基材料的常见手段,被人们普遍使用[57]。Yang等[58]成功制备定量N 掺杂的吡啶-石墨炔(PYGDY) 和嘧啶-石墨炔(PM-GDY) 大尺寸薄膜。PY-GDY 和PM-GDY 薄膜均匀连续、透明、可弯曲,均可直接用做锂离子电池的负极材料。PYGDY 和PM-GDY 不但保留本征GDY 均匀分布的六方大孔分子结构,而且N 的掺入有效增大了层间距,这有利于Li+的扩散及嵌入和脱嵌。刘辉彪等[59]也基于超分子化学的新方法对GDY 进行了原位N 掺杂,利用GDY 和有机共轭分子间的强π-π 相互作用,在铜片表面原位制备了石墨炔/卟吩复合薄膜。将其直接用作锂离子电池负极材料,比容量提高到了1 000 mAh·g−1,该复合电极也表现出优良的倍率性能和循环稳定性。
使用常规的杂原子掺杂方法,掺杂位置常不可控,这阻碍了对掺杂态GDY 构效关系的深入理解,也限制了对其催化活性的改性提升效果。王丹团队[60]发明一种通过周环反应对GDY 进行sp-N 精准掺杂的方法。研究其ORR 性能发现,相比于其他N 原子掺杂构型,sp-N 掺杂使得相邻的碳原子带有更多的正电荷,更有利于O2的吸附和活化,同时N 的掺杂可加快催化剂界面的电子转移。和传统掺N 碳基催化剂的吡啶N 被认为是ORR 活性中心不同,该材料中sp-N 被认为是反应活性中心,随着sp-N 含量的增加,催化活性提高。这种sp-N 原子的可控掺杂方式,可能为其他富炔键材料的定点掺杂提供借鉴。与单一杂原子掺杂相比,多种杂原子掺杂对提高炭材料OER 性能方面具有更大的优势。在一般情况下,S 原子优先取代苯环上的碳原子,而N 原子掺杂位点却有多种可能,多位于缺陷和边缘位置处。由于N、S 原子的相对距离不可控,因此在实验上难以对N、S 原子的协同效应进行定量评估。针对这一难题,该课题组在上述研究工作的基础上开展了sp-N 原子和S 原子共掺杂对GDY 的OER 性能的影响研究[61]。研究人员利用三聚氰胺和二苄基硫分别作为N、S 掺杂源,制备了掺杂位点明确的sp-N、S 共掺杂GDY。sp-N 原子在GDY 中的掺杂位置确定,而S 原子取代苯环碳,从而实现N、S 原子的间距可控,N 与S 之间的距离在不超过0.75 nm 时可以获得较强的协同效应。sp-N 是所有掺N 构型中最优选择,在sp-N 掺杂的基础上再掺杂S,能使OER 电流密度进一步提高。sp-N、S 共掺杂大幅降低GDY 的界面电阻和电荷转移电阻,OER 活性不仅高于N 或S 单掺杂的GDY,还高于经典的RuO2催化剂,证明N、S 原子掺杂对提升GDY 的OER 性能方面表现出显著的协同效应。
3.2 金属原子改性
GDY 也是金属原子的理想载体,GDY 能够通过自身结构C≡C 中的π 键与金属原子d 轨道相互作用来锚定金属原子并形成很强的结合力,而GDY 中均匀的多碳原子三角炔基大环则为锚定金属原子提供了理想空间,在其中锚定金属原子后,金属原子与GDY 之间会发生强烈的电荷转移。Ma 等[62]选择了GDY 上6 种金属原子潜在的吸附位点(图7a):即六元芳环中心上方的空位(H1)、三角乙炔环中心上方的空位(H2)、sp 和sp2杂化碳原子间的桥位(B1)、两个sp 杂化碳原子间的桥位(B2)、两个sp2杂化碳原子间桥位(B3)、以及乙炔环的角位(C),利用第一性原理计算比较了单原子(SAs)贵金属(Au、Pt、Ir、Pd、Rh、Ru)在6 种不同位点上的吸附能。研究发现,GDY 上贵金属SAs 的最优吸附位点是三角乙炔环中心上方(H2),不同贵金属原子与GDY 的结合强度由大到小排序为:Ir > Ru > Pt > Rh >Pd > Au。将GDY、石墨烯与金属原子的结合能力对比发现,贵金属原子在GDY 上的吸附能远大于石墨烯上。这是因为石墨烯薄片中只有sp2杂化的碳原子,垂直于石墨烯平面的方向上只有面外Pzπ/π*轨道存在,而GDY 上中存在活跃的sp 杂化碳,这种杂化使得面内Px-Pyπ/π*轨道可在垂直于C≡C 键的方向任意旋转,因此贵金属原子的价电子不仅可以与Pz轨道杂化,还可以GDY 碳原子的Px、Py轨道杂化,炔环上的π/π*轨道始终指向贵金属原子,导致贵金属原子与GDY 的相互作用更强,证明GDY 可以作为金属SAs 催化剂的良好载体。另外,使用过渡金属原子对GDY 进行掺杂时,二者之间的相互作用会导致过渡金属原子的s、p 和d 轨道电子重新分布,对GDY 的电子结构及物化性能具有良好的调控作用。Liu 等[63]利用模拟计算研究了过渡金属SAs 催化剂Ni-GDY 和Cu-GDY 的性能。首先探究了Ni 和Cu 在H1,H2 和C 等3 个不同GDY 嵌入位点上的稳定性差异(图7b)。结果表明在所有的嵌入位点中,炔环角位点C 处过渡金属原子和碳原子之间的成键键长最短,结合作用最强。由于Ni、Cu 原子与碳原子的电负性差异较大,它们嵌入后会引起GDY 上电荷的重新分布。Ni 和Cu 原子显正电性,分别给GDY 提供0.271 e−和0.272 e−,导致GDY 费米能级上移,整个TDOS 向较低能级移动(图7c-d),功函数减小。和过渡金属不同,低电离势碱金属原子(AM=Li,Na,K)易与GDY 的十八元六边形环相互作用,其高度离域的大π 共轭结构可以电子传输的良好通道,可形成分子内电子供体-受体框架结构(D-π-A),计算证明碱金属的掺杂改变GDY 的电子结构后有助于提升其非线性光学性质。AM@GDY 的垂直电离势低于碱金属AM,而复合后静态超极化率大幅增加,其中K3@GDY的静态超极化率最大,达到1.61×105a.u[64]。
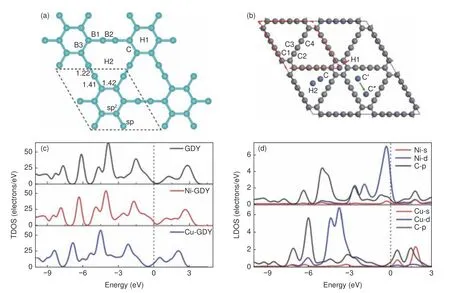
图7 (a) 单原子贵金属(Au、Pt、Ir、Pd、Rh、Ru)吸附位点[62];(b) Ni、Cu 在GDY 上可能嵌入的三个位点;(c) 本征GDY 和在C 位点上嵌有Ni 和Cu 原子时的TDOS;(d) 掺杂金属原子s 和d 轨道相邻四个碳原子p 轨道的LDOS 投影[63]Fig.7 (a) Adsorption sites of monatomic precious metals (Au, Pt, Ir, Pd, Rh, Ru)[62]; (b) Three possible sites of Ni and Cu in GDY; (c) Intrinsic GDY and TDOS with Ni and Cu atoms embedded at the c site; (d) LDOS projections of the p orbitals of four carbon atoms adjacent to the S and d orbitals of the doped metal atoms[63].Reprinted with permission
超碱金属团簇通常具有比碱金属原子更低的电离势,更容易释放价电子,掺杂在GDY 中能让复合体获得过量电子。Shehzadi 等[65]基于DFT 计算理论,设计了一系列超碱金属掺杂GDY 的M2X@GDY (M = Li,Na,K 和X = F,Cl,Br)复合物。计算证明这些超碱金属容易稳定键合在GDY 的大三角炔环位点上,超碱金属团簇的掺杂减小了HOMO-LUMO 能隙,从而改善了GDY 的导电特性。计算GDY 到超碱金属团簇的核间距离发现,核间距离随着超碱金属原子尺寸的增大而增大。核间距离越大,M2X 与GDY 之间的相互作用越弱,M2X@GDY 复合物的稳定性越低。通过理论计算指导GDY 的掺杂改性,有助于GDY 基功能材料在众多领域的广泛应用。
如上述众多理论计算表明,GDY 的18 碳大炔环结构可以为单金属原子提供理想的锚定位点,形成稳定的SAs 催化剂。鲁统部团队[66]利用湿化学法,将GDY 与K2PtCl4溶液在273 K 下反应8 h,直接获得催化剂Pt-GDY1。再将Pt-GDY1 置于Ar 气氛中,473K 退火处理1 h,得到催化剂Pt-GDY2。Pt 原子与炔基C 原子之间强的相互作用使其均匀分散在GDY 上,Pt-GDY1中形成C1-Pt-Cl4五配位物种,Pt-GDY2 中形成C2-Pt-Cl2四配位物种,两种SAs 催化剂均保持GDY 的形貌,没有形成明显的Pt 团簇。Pt 原子的配位环境直接影响SAs 催化剂的电催化析氢反应(HER)催化活性,Pt-GDY2 的质量活性分别是Pt-GDY1 和Pt/C 的3.3 倍和26.9 倍,这是因为Pt-GDY2 中Pt 的5d 轨道具有更高的未占据态密度,且Pt 活性位的氢吸附自由能近等于零。该实验研究进一步证明,GDY 高的比表面积不但有助于金属SAs 分散,而且分子中的炔基碳与金属原子之间的强配位作用明显提高了SAs 催化剂的稳定性,是一种优异的金属SAs 催化剂载体材料。Xue 等[67]通过电化学还原法将Ni、Fe 金属原子沉积在多孔GDY 泡沫上,合成了GDY 基过渡金属(Fe、Ni)SAs 催化剂,其中Ni、Fe 负载质量分数分别为0.278%和0.680%,且金属原子均显零价。通过计算GDY 与Ni/Fe 原子之间的结合能可以确定最佳掺杂构型,Ni/Fe 原子在GDY 上炔键位处(−3.72/−1.22 eV)比在苯环位(1.39/0.37 eV)结合能更强,所以前者是优先吸附位点。化学键键长也间接反映原子间相互作用强弱,炔键处Ni/Fe―C 键键长更短,结合更稳定,并且Ni/Fe 向GDY 存在较强的电荷转移,这显著提高了SAs 催化剂的导电性和催化活性。另外,Raman 分析D带与G带峰强度比值(ID/IG)发现,Ni/GDY(0.87)和Fe/GDY(0.85)的均大于GDY(0.77),表明Ni/GDY 中形成了更多的缺陷和活性位点,从而提高了HER 催化效率。
3.3 金属、非金属原子共改性
钴掺杂的炭材料往往展示出优异的ORR 和HER 性能,是一类廉价易得的电催化材料。然而,如何增强钴和碳原子之间的相互作用以提高钴掺杂GDY 的稳定性和钴的有效掺杂量是一大难题。Wang 等[68]合成了钴-氮共掺杂石墨炔(Co-N-GDY)。1% Co-N-GDY 催化剂的微观照片呈现三维结构和粗糙微观表面,其中明显的孔隙结构使其拥有丰富的缺陷和活性位点。用XPS 测定了1% Co-N-GDY 表面的Co 和N 原子百分数,分别为0.47%和5.39%。可见,尽管在合成过程中只使用了少量的钴盐前驱体,但GDY 网络中Co 原子的掺杂量较高,这是由于Co 与GDY 之间存在较强的相互作用,GDY 中sp 和sp2杂化结构更有利于Co 原子的锚定,这也赋予了Co-N-GDY 良好的稳定性。N 和C 原子的原子半径接近,但N 原子具有更高的电负性,N 掺杂可以改变GDY 的电子构型,而Co 原子的引入进一步增加了N-GDY 的反应活性位点。因此,Co、N 原子的协同作用赋予了Co-N-GDY 优异的ORR 和HER 催化性能。
掺杂能够调变GDY 的电子结构,破坏其分子结构的有序度和对称性,产生晶体缺陷及不饱和的碳原子,从而改变其物理化学性质和表界面性能。但向GDY 引入少量非金属原子时更倾向于掺杂,而引入金属原子时,需要严格控制剂量,只有少量的金属原子,才可能和GDY 形成单原子配合物。如果金属原子或离子过量,很可能形成GDY 和金属单质颗粒或者金属氧化物颗粒的复合物,不能达到掺杂改性的目的。
3.4 基团修饰改性
刘辉彪等[69]运用Huisgen 环加成实现了叠氮烷基化修饰G D Y,所合成的G D Y-T z-CH2(CH2)xCH3(x= 4,10,16)在众多有机溶剂中具有良好的分散性,解决了其在电子器件应用中分散性性和加工性差的问题。将G D Y-T z-CH2(CH2)16CH3应用于电子传输层的平面异质结钙钛矿太阳能电池中,将同类太阳能电池的能量转化效率从16.24%提高到19.26%,短路电流从20.85 增大到23.70 mA cm−2。他们还使用1-溴芘与GDY 发生Sonogashira 交叉偶联反应合成了GDY-Py 量子点(GDY-Py QDs),从而得到有良好分散性能的荧光材料[70]。GDY-Py QDs 平均直径为3 nm 左右,在多种有机溶剂和水中的分散性较GDY 都有所提高。GDY-Py QDs 的共轭结构有利于提高其电子转移效率,相对量子产率高达42.82%,而且作为造影剂在细胞成像中性能优异。
4 石墨炔的应用
4.1 石墨炔在环境领域的应用
4.1.1 环境催化
环境催化是环境保护和绿色化学重要的科学与技术基础,也是催化领域发展最为迅速,成就最为显著的学科方向之一。其实质是利用催化反应实现污染控制,使污染物减量化或无害化转化,以达到改善和保护生态环境的目的。
近年来,由于异质界面特殊的电荷转移效应,光催化剂与炭材料复合是提升光催化效率的有效手段之一。GDY 的本征带隙为0.46 eV,电导率为2.56 × 10−1S m−1,具有半导体特性和较大的载流子迁移率,可作为光电催化剂的新型载体,增强水体中的有机污染物的去除。Thangavel等[71]在碱性条件下使用水热反应制得石墨炔-氧化锌(GDY-ZnO)复合光催化剂,并用于光催化降解两种常见的偶氮染料(亚甲基蓝和罗丹明B,模拟废水体积100 mL,污染物浓度1×10−3M,催化剂固含量5 mg L−1),结果表明GDY-ZnO 的光催化活性高于ZnO,GDY 的复合使ZnO 对两种染料污染物的降解速率增大了近一倍。在GDYZnO 复合光催化剂中,GDY 中的二乙炔键可能产生杂质能带,致使价带上的电子更容易被激发并跃迁到导带,最终提高了光催化反应效率。Dong 等[72]利用氮掺杂锐钛矿型TiO2纳米片与GDY 复合制得GDY-NTNS 光催化剂,并研究其在可见光照射条件下降解罗丹明B 的催化活性(模拟废水体积40 mL,污染物浓度10 mg L−1的,催化剂固含量25 mg)。相比NTNS,反应240 min时,GDY-NTNS 光催化罗丹明B 的去除率从78%提高到约90%。GDY-NTNS 的禁带宽度约为3.08 eV,可见光照射下电子从TiO2价带(VB)被激发到导带(CB)上,并在VB 中留下空穴。由于TiO2的CB 电位(0.47 eV)大于GDY 的费米能级(0.33 eV),而GDY 具有较高的导电性,TiO2中CB 的电子可以向GDY 移动,并沿GDY 界面转移。吸附在GDY 上的O2接受电子,生成·O2−。h+和·O2−最终对降解罗丹明B 氧化降解。氮掺杂增强了TiO2对可见光的吸收,GDY 的引入提高了GDY-NTNS 光生电子-空穴对的分离效率。Lin 等[73]合成了具有良好光催化活性和稳定性的新型光催化剂Ag3PO4@γ-GDY,用于抗生素等难降解有机废水的处理(模拟废水体积100 mL,污染物浓度20 mg L−1,催化剂固含量50 mg)。具有较高载流子输运能力的γ-GDY 可以作为光生电子的有效传输层,它迅速将Ag3PO4价带上被激发的电子输送到光催化剂表面。分别在5、8 和16 min 内,对萘酚、诺氟沙星和苯酚的去除率达到100%。
在机械力的作用下,GDY 可能会发生不规则的变形,导致内部电荷迁移,从而表现出一定的压电响应。Huo 等[74]采用球磨法制备了具有压电-电催化性能的MoS2/GDY,并以四环素为目标污染物考察其降解性能。优化后的MoS2/GDY压电响应比MoS2高35.29%,具有良好的催化活性,球磨触发压电催化40 min 后对四环素的降解率达到92.15%。在GDY 框架中掺入杂原子,可以改变其对称性以获得更大的压电响应,同时,产生额外的活性缺陷位点有助于提高其催化活性,如Zhang 等[75]合成了一种可用于净水的硫掺杂石墨炔(SGDY)纳米片模拟酶催化剂,通过超声激发压电增强的SGDY 可高效降解染料和抗生素。S 的掺杂使GDY 带隙减小,改善了其网络的电荷分布,增加了催化活性位点的数量,使它具有明显的类过氧化物酶活性。同时,硫原子的加入打破了GDY 的反转对称性并构建内置电场。在内置电场的推动下,载流子向纳米片表面移动,参与氧化还原反应,导致SGDY 催化H2O2分解活性显著增加。
可见,GDY 独特的电子结构和良好的载流子传输能力,对其复合催化剂光催化性能的提升发挥着关键作用。而且GDY 的能带结构可通过掺杂或者改性修饰进行有效调控,这就为其拓展光催化应用领域提供可能。
4.1.2 CO2光/电催化还原
化石燃料的日益短缺和CO2的过量排放,导致能源环境问题日趋突出[76]。利用光/电催化将CO2转化为低碳化学品是缓解上述难题的潜在方案之一。GDY 作为助催化剂可用于CO2还原,助力温室气体控制。余家国团队[77]将Zeta 电位为负的GDY 与带正电荷的TiO2在水溶液中进行静电自组装,合成了TiO2纳米纤维和GDY 复合的异质结TiO2/GDY。将其用于光催化CO2还原,生成产物包括CO、CH4和O2,如图8a 所示,其中C O 为主要产物,生成速率最高可达5 0.5 3 μmol h−1g−1。GDY 高能乙炔键中的电子容易离域以达到最稳定状态,被认为是吸附和催化CO2的活性中心。作者们计算TiO2和GDY 静电势,从真空能级和费米能级的能差得到材料的功函数(图8b,c),可知TiO2(101)晶面比GDY(001)晶面具有更大的功函数(5.71 eV vs.5.13 eV)。当GDY 与TiO2复合后,GDY 中的离域电子与TiO2中的Ti 3d 空轨道发生杂化,电子将会从GDY 流向TiO2以达到相同的费米能级。图8d为TiO2/GDY 异质结构的电荷密度差侧视图,青色区域和黄色区域分别表示电子的消耗和积累,揭示界面处GDY 向TiO2转移0.021 e−,该电子转移致使TiO2/GDY 界面处形成了内建电场。在光照时,TiO2的光生电子在内建电场驱动下会定向快速转移到GDY 纳米片上,富电子的GDY 纳米片充当电子“洼地”的角色,同时GDY 对CO2分子有很强的化学吸附和活化能力,保证了CO2分子高效催化还原成为CO 和CH4(图8e)。此外,GDY 的光热效应引起局部加热,也加速了光催化还原反应过程。该研究小组[78]还使用含二甲亚砜的溶剂热法制备了GDY 修饰的CdS 纳米晶光催化剂(CdGDY),直接研究其对CO2气体的催化还原性能。与石墨烯修饰的CdS 纳米晶光催化剂(CdG)相比,CdGDY 具有更高的活性、选择性和稳定性。同时,GDY 和石墨烯的引入均可促进CO2在催化剂表面的吸附,而CdGDY 的吸附能力远远强于CdG 和CdS,CdGDY 和CdG 上的C O2光催化还原转化速率分别为1 8.7 2 和14.97 μmol h–1g–1。研究认为,CdGDY 中GDY 和CdS 界面处形成了稳定的Cd―O―C 化学键,该键诱导CdS 中形成S 空位和GDY 中双炔键的缺电子状态,共同增加了CdGDY 表面CO2的吸附位点。空位等丰富的缺陷有利于降低反应能垒,将吸附态的CO2活化转化为重要中间体CO2−,接着被进一步还原为CO,CH4及CH3OH 等产物。根据差分电荷密度结果,CdGDY 和CdG 中电子均倾向于穿过异质结界面从CdS 流入GDY 或GO,但前者这种趋势更强。因此,在强界面相互作用、快速的电子定向传导及稳定的电子储存能力协同作用下,CdGDY 展示出优异的光催化还原CO2性能。
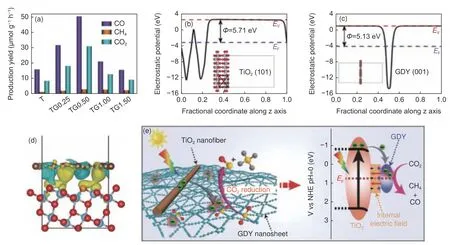
图8 (a) 不同负载量时TiO2/GDY 的CO2 还原光催化活性;(b) TiO2 的(101)面和(c) GDY 的静电势;(d) TiO2/GDY 电荷密度差的侧视图;(e) CO2 光还原中TiO2/GDY 在紫外光照射下内部电场诱导电荷转移和分离示意图[77]Fig.8 (a) Photocatalytic activity of TiO2/GDY for CO2 reduction with different loading capacity; (b) TiO2(101) surface and (c)GDY electrostatic potential;(d) Side view of TiO2/GDY charge density difference; (e) Schematic illustration of TiO2/GDY heterojunction: internal electric field-induced charge transfer and separation under UV–visible light irradiation for CO2 photoreduction[77].Reprinted with permission
如前文所述,GDY 独特的结构可以有效地稳定SAs,并促进GDY 与原子中心之间的电荷转移导致单个原子的电子环境发生变化。Shi 等[79]将GDY 作为基底材料,在CuSAs 和GDY 之间构建Cu-C 配位键来调控Cu SAs 的电子环境。Cu SAs/GDY 不仅可以稳定Cu SAs,Cu-C 键为CO2-CH4的8 电子还原过程提供一个有效的电荷转移通道,还可以促进Cu SAs 上中间体*OCHO 的高选择性生成,阻止CO 或其他多碳产物的出现,使反应途径更容易生成CH4。黄勃龙等[80]计算表明GDY 稳定的镧系单原子催化剂(GDY-Ln SAC)在CO2还原制取合成气方面具有优势。镧系金属的f 轨道和GDY 中烷基p 轨道的分别对CO2还原和HER 中的反应物CO2和H2O 的吸附起到关键作用,通过对GDY-Ln SAC 上多活性位点的调控能够实现合成气中CO/H2比例的灵活调变。
另外,GDY 还有助于金属纳米催化剂选择性地将CO2还原为多碳(C2+)产物,Chang 等[81]在芘基石墨炔(Pyr-GDY)纳米纤维上原位生长了高度分散的超小(~2 nm)Cu 纳米粒子,获得电催化剂Pyr-GDY-Cu。Pyr-GDY-Cu 电催化CO2还原的C2+产物包括C2H4、C2H5OH 和C2H7OH 等,法拉第效率(FE)高达74%,显著高于无载体Cu 纳米颗粒(C2+FE,~2%)、碳纳米管负载Cu 纳米颗粒(CNT-Cu,C2+FE,~18%)、GO 负载铜纳米颗粒(GO-Cu,C2+FE,~8%)和其他报道的超细铜纳米颗粒催化剂。通过炔键与Cu 纳米粒子的强配位作用,更多的电子从超细Cu 纳米粒子流向Pyr-GDY 基底,降低了超细Cu NPs 上H+对H*的还原能力,从而抑制析氢反应。Pyr-GDY-Cu 和Cu-NP 的d 带中心计算结果为−3.74 和−3.03 eV。与Cu 纳米粒子相比,Pyr-GDY-Cu 的d 带中心发生了明显的位移。d 带中心影响其和吸附质之间的相互作用,d 带中心越低(相对于费米能级)说明催化剂和吸附质之间的相互作用越弱。通过调整催化剂的d 带中心,Pyr-GDY 载体削弱了Cu 纳米颗粒与*H 和*CO 中间体之间的结合,从而减少C1产物生成,而强化了*C O 之间的C―C 耦合过程,最终提高了C2+产物的选择性。另外,Pyr-GDY 作为载体时,炔基基团和Cu 纳米粒子之间的强相互作用有效阻止了Cu 纳米颗粒在电催化过程中的团聚,提高了催化剂的稳定性。GDY 对Cu 催化CO2电还原选择性具有关键影响,证明GDY 作为载体材料在电催化CO2转化领域拥有广阔的研究前景。
4.1.3 污染物检测
某些痕量的重金属离子或者抗生素尽管其浓度不高,但若在人体内长期累积,会与含有N、S、O 的生物配体结合,引发严重的健康问题。GDY 拥有较高的比表面积和良好的导电性,有利于电化学分析检测过程中被检测物质的预富集和电荷的转移,可以用作电分析基体材料[82–84]。
最近,孙宝云团队等[85]构制了一种GDY 修饰的玻碳电极GDY/GCEs(图9a),利用电化学预富集和阳极溶出伏安法检测实际水样中Cd2+和Pb2+。以石墨烯为对比样,研究了炔键对检测Cd2+/Pb2+的影响。Cd2+或Pb2+吸附后,GDY 的XPS 图谱出现了新的峰(图9b),该轨道结合能降低。富含π 电子的炔键基团能给GDY 提供电子,由于Cd2+和Pb2+可提供空轨道接受GDY 的s p 杂化碳的部分电子,从而金属阳离子被GDY 吸附后趋向于向低价态转化,有利于更多的金属阳离子的定向迁移。同时,与石墨烯相比,GDY 的炔键可以提供电子与重金属离子相互作用并充当吸附活性位,而石墨烯表面吸附位点相比较少,通常需要修饰或复合来增加吸附位点。如图9c-e 所示,GDY 电化学传感器在两种重金属离子检测过程中展示出较宽的线性范围、低的检出限和较好的重现性。在大多数其他干扰离子(如Cu2+、Ni2+、Zn2+、Hg2+、SO42-、Fe2+、NO3−、K+等)存在的情况下,GDY/GCEs 对Cd2+和Pb2+具有良好的选择性。随后,他们又合成了精准吡啶N 取代碳原子的三嗪类石墨炔(TGDY),将其修饰在玻碳电极上构筑电化学传感器,同样用于水中Cd2+、Pb2+的分析[86]。炔键和吡啶N 与Cd2+、Pb2+之间具有适宜强度的亲和作用,其中TGDY 提供电子,Cd2+或Pb2+提供空轨道,二者之间产生螯合吸附,因此,基于TGDY 的电化学传感器显示出优异的电分析性能。干扰离子对目标离子的干扰较小,通过掩蔽剂可将干扰离子(如Cu2+或Ni2+)的影响降至最低。该工作明确了双炔键和吡啶N 原子都是较好的电化学分析活性位点,可控合成具有特定类型和数量的活性中心的GDY 电极材料将有助于开发高效的环境分析传感器。可见,吡啶N 对GDY 掺杂不但改善了传感器的电化学性能,而且作为活性位点提高了Cd2+、Pb2+检测的灵敏度。
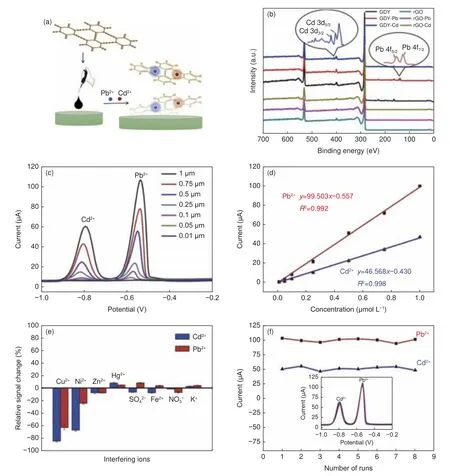
图9 (a) GDY/GCE 实现重金属离子检测的示意图;(b) GDY 和rGO 吸附Cd2+或Pb2+前后的XPS 光谱;(c) Cd2+和Pb2+的电化学响应及(d)相应的线性关系;(e) GDY/GCE 在5 μM 干扰离子存在下的选择性;(f) GDY/GCE 的重现性[85]Fig.9 (a) Schematic diagram of detection of heavy metal ions by GDY/GCE; (b) XPS spectra before and after adsorption of Cd2+ or Pb2+ by GDY and rGO;(c) Electrochemical responses of Cd2+ and Pb2+ and (d) corresponding calibration curves; (e) Selectivity of GDY/GCE in the presence of 5 μM interfering ions;(f) Reproducibility of GDY/GCE[85].Reprinted with permission
双酚A(BPA)是一种重要的内分泌干扰物,有致癌性、神经毒性,可能引起心血管疾病、导致不孕不育、影响人们的正常生长发育。BPA 已在人类体液、胎盘和胎儿肝组织中普遍检出,鉴于BPA 的高暴露率及其对人类健康的不利影响,迫切需要开发可靠、高效的BPA 检测分析方法。卢宪波等[87]将GDY 作为酪氨酸酶固定化的稳定基质,构建了基于GDY 的酪氨酸酶电化学生物传感器(Tyr-GDY-Chi/GC),用于BPA 的超灵敏检测。该传感器在1.0×10−7到3.5×10−6mol L−1的BPA 浓度范围内呈线性响应,灵敏度高达2 990.8 mA cm−2M−1,检测限低至24 nmol L−1。在BPA 检测方面,Tyr-GDY-Chi/GC 表现出比碳纳米管基和石墨烯基酪氨酸酶电化学生物传感器更好的分析性能。这归因于GDY 和BPA 之间的强 π−π 相互作用,这有助于BPA 在传感器上的快速富集并与酪氨酸酶反应。随后,他们在NH3气氛中煅烧GDY 合成掺N 石墨二炔(NGDY),并以NGDY 为生物传感平台,成功制备了基于乙酰胆碱酯酶的农药生物传感器和基于酪氨酸的苯酚生物传感器[88]。NGDY 具有较大的比表面积、分级纳米多孔结构、高结构缺陷和良好的导电性,有利于酶的固定化、物质扩散和电子传输,NGDY 中存在不同构型的N 原子(石墨N、亚胺N 和吡啶N)在提高电化学生物传感器性能方面起着关键作用,石墨N 增加了GDY 的电导率,而亚胺N 和吡啶N 有利于吸附和富集目标污染物分子。这两种酶电化学生物传感器的灵敏度高,重现性和稳定性好,可以作为环境中酚类化合物快速检测的有力工具。
另外,GDY 也可以用做环境中低浓度抗生素的分析材料。吴萍等[89]合成了sp―N 掺杂石墨炔量子点(NGDYQDs),发现它可以用于氯霉素的定量电化学分析。sp―N 掺杂和量子尺寸效应改变了GDY 的电子特性,NGDYQD 对氯霉素中的―NO2基团还原为―NHOH 表现出高的电化学活性。所得的电化学传感器对氯霉素的线性响应范围为0.1 到114.5 μmol L−1,检测限约为5 nmol L−1,灵敏度为8.79 μA−1μM−1cm−2,同样展示出高的重复性、再现性和稳定性。可见,GDY作为一种新型二维全碳纳米材料,可以提供一种高效的传感器平台,用于电化学传感器及生物传感器的构制,在环境污染物检测和生物医学分析方面有很好的用途。
4.1.4 吸附分离
随着全球范围核电站数量的持续增长,核设施产生的放射性惰性气体如Xe、Kr 对生态环境和人类健康有极大的危害。核设施产生的85Kr 的半衰期约为11 年,而127Xe 半衰期较短,但其产量是Kr 的10 倍左右。为了削减需要长期储存的放射性废气的体积并实现Xe 的回收再利用,人们设想借助吸附的方法将Xe 从Xe、Kr 混合废气从分离出来。常见的Xe 和Kr 吸附分离材料为多孔材料,如活性炭、沸石和多孔聚合物等[90,91],GDY 的孔洞结构和较大的比表面积使其在吸附分离方面呈现一定潜力[92–94]。Vazhappilly 等[95]通过理论计算分别研究了本征GDY 和掺杂GDY 材料(Be、B、N、Al、Si 和P 等元素掺杂)对放射性惰性气体Xe、Kr 的吸附分离性能。因掺杂原子与碳原子的电负性和原子半径的差异,掺入杂原子后会引起GDY 上电荷密度的重排和晶格的重构,从而影响它的吸附性能。研究发现,本征GDY 对惰性气体Xe 和Kr 的吸附作用不强,但掺杂GDY 对Xe、Kr 气体吸附作用显著增强,其中p-型Al 掺杂的GDY 为最佳吸附剂,吸附性能与沸石、MOFs 等多孔材料相当,这和Al 原子向GDY 上C 原子强烈的电子转移导致其缺电子和掺Al 后导致严重的晶格畸变有关。与Kr 相比,Xe 的原子极化率更高,掺杂GDY 对Xe 的吸附力更强。Zhou 等[96]制备了一种多层GDY 纳米多孔气体分离膜,该膜允许氦和氢等轻气体快速穿透,而对氙等重惰性气体则表现出较强的抑流作用。
除此之外,石油、化学品泄漏事故也是造成环境污染的重要事件之一。以GDY 作为吸附材料从水中收集溢出的油或有机溶剂是一种操作简单、效益高、速度快的方法[97–99]。Liu 等[100]将GDY 薄膜附着在三聚氰胺海绵骨架上,利用GDY 的疏水亲油性质,合成了一种能够有效收集油污的GDY 海绵(GDYMS)。GDYMS 具有三维多孔结构、疏水性好、质量轻、稳定性好等特点,其纯水的接触角约为132°。GDYMS 可以高吸附容量选择性地吸附废水中高达自身重量160 倍的油和有机溶剂。此外,由于GDY 的非凡稳定性,GDYMS 在腐蚀性液体(有机溶剂、强酸、碱性溶液)中表现出很高的吸附效率和高回收率,无论是浮在水面上还是沉在水面下,都能很好地吸油和各种不同密度的有机液体(如汽油、CHCl3)。此外,GDYMS 海绵可以重复使用,吸附的油和有机溶剂也都可以通过挤压回收,表现出良好的可循环利用性能(>100 次循环)。GDY 对各种油类和有机溶剂具有良好的油水分离效果,可以用做处理石油和工业化学试剂泄漏的新型吸附分离材料。
4.2 石墨炔在能源领域的应用
4.2.1 二次电池
炭材料因具有良好的导电性、优良的电子传导和离子输运能力,是最常见的电极材料。GDY 及其改性、复合材料作为负极材料,在二次离子电池中的电化学性能研究最先吸引研究人员的兴趣。锂离子电池(LIB)是目前最成熟的商用二次电池,其输出电压较高,能量密度大,循环使用寿命长,已被广泛应用于移动电子设备、电动汽车、军事装备等。黄长水等[29]在铜表面生长不同厚度的GDY 直接作为负极材料,研究其锂离子电池的性能。研究发现该电池具有高比容量、出色的倍率性能和长的循环寿命。在500 mA g−1的电流密度下,循环 400 次后,可逆比容量高达520 mAh g−1;在2 A g−1的高电流密度下,1 000 次循环后保持其高的比容量420 mAh g−1,负极上多层GDY 储锂主要是通过层间嵌入/脱出过程和表面吸收/解吸过程共同实现。随后他们又研究发现,对原位生长的GDY 膜进行Ar 保护煅烧处理,也可以进一步提高其锂电性能[30]。400 °C 热处理下的GDY 薄膜的储锂性能好、可逆容量最高,优于石墨烯、碳纳米管、N 掺杂多孔碳纤维及天然石墨等其它炭材料,组装LIB 的库伦效率达到98.2%。200 和300 °C 热处理的样品可逆容量较低,可能是GDY 表面存在低导电性的低聚物和颗粒,限制了Li+活性结合位点。随着热处理温度升高,GDY 膜上低聚物不断消除,导致GDY 能隙减小和电导率增加。但经500 °C 处理的样品,虽然GDY 导电性变好,但不利于Li+的扩散,致使可逆容量衰减和倍率性能下降。
在铜片催化原位生长合成层状GDY 薄膜的基础上,黄长水团队[101]又以CuO 纳米球为模板和催化剂,使用溶剂热法制备了具有高表体比的石墨炔纳米空心球(GDY-HNSs)。通过调节前驱体的量,能够控制合成不同壳层厚度(4 0 ~200 nm)的GDY-HNSs(图10a-d),并研究其锂电性能。GDY-HNSs-120 优异的电化学性能与其独特结构有关,如图10f 所示,首先,空心纳米球表面纳米壁的层次结构赋予了GDY-HNSs-120 高的比表面积、丰富的电化学活性中心和大的电解液接触面积;其次,空心结构中可穿透的壳层有利于离子和电子的快速扩散;最后,GDY-HNSs 壳层的纳米孔(包括微孔和中孔)以及GDY 结构中的边缘和空位等缺陷可以提供更多的储锂位点,有助于增大可逆容量。同时,这些空心纳米球壳层的纳米孔道和中空腔体为Li+重复嵌入/脱出引起的结构应变和可能的体积变化提供足够的空间。
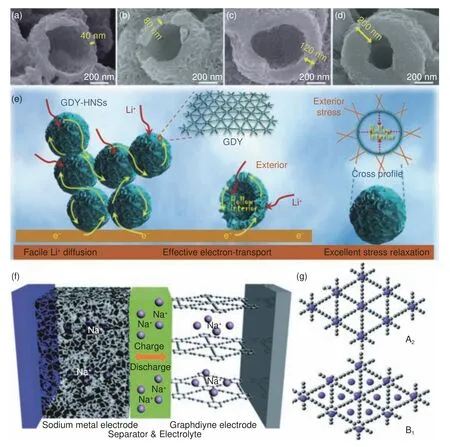
图10 (a-d) 不同壳层厚度GDY-HNSs 的SEM 图像;(e) GDY-HNSs 阳极的锂离子电池原理图;(f) GDY-HNSs 电子传输、锂离子扩散和应力松弛的示意图[101];(g) Na+-GDY 的可能构型[104]Fig.10 (a-d) SEM images of GDY-HNSS with different shell thickness;(e) Schematic diagram of lithium-ion battery with GDY-HNSS anode;(f) Schematic diagram of electron transport, lithium ion diffusion and stress relaxation in GDY-HNSs[101];(g) Possible configurations of the Na+-GDY[104].Reprinted with permission
对炭电极进行N 掺杂改性是提高LIB 电化学性能的有效措施,N 原子掺杂不仅可以调节炭材料的电子性质,还能够改变其表面极性。然而,GDY 中特定构型N 的准确掺杂一直是研究难题。肖胜雄小组[102]报道了一种水热法高效原位合成高质量吡嗪喹喔啉基石墨二炔(PQ-GDY)薄膜的策略,这种自上而下的方法实现了氮原子掺杂的精准控制。作者们就PQ-GDY 的层状堆叠模式、锂结合能力和储锂性能开展了详细研究。DFT 计算对比了PQ-GDY 层间AA 型堆叠、6 种不同AB 型堆叠及2 种ABC 型堆叠方式的能量差异,认为AB-6 型堆叠方式能量最小,是多层PQ-GDY 薄膜最可能的存在形式。理论层间距为0.330 5 nm,这和HRTEM 和XRD 计算所得层间距结果(分别为0.33 和0.331 nm)非常吻合。与以单苯环或单杂环为中心的GDY 相比,PQGDY 中交联的N 杂芳香环不仅增大了电子离域程度,还为Li+提供了稳定的结合位点。通过半电池测试和DFT 计算证明,插层过程中Li+与PQGDY 负极上3 种不同活性位点之间的亲和作用大小顺序为吡嗪N>双炔碳>中心芳环。在电流密度200 mA g−1时,经800 次循环后,可逆比容量为570.0 mA h g−1,达到自身理论容量的97.2%,半电池的平均库伦效率为98.48%。
目前,各行各业对二次电池的需求日益增加,而全球锂资源供给短缺,因此,人们亟待开发锂电池的替代产品。元素钠、钾和锂的化学相似,储量丰富,钠、钾离子电池可能作为锂电池的有效补充。Na 比Li 更便宜,含量也更丰富,GDY 为Na 提供了较大的孔隙和空间,Na 可以插入并贯穿其中。Farokh Niaei 等[103]利用DFT 计算探究了GDY 作为钠离子电池负极材料的理论容量。计算所知,在不考虑GDY 单元电池膨胀的情况下,单个GDY 层对Na+的最大容量为NaC2.57(相当于497 mA h g−1),GDY 堆积层的最大容量为NaC5.14(相当于316 mA h g−1)。因此,将Na+插入GDY 作为可充电电池的负极材料具有一定的潜力。Zhang 等[104]合成具有微/介孔结构的GDY 粉末,并将其作为钠离子电池负极材料(图10g)。由于该GDY 的三维多孔结构、化学稳定性和较好的导电性,组装的钠离子电池显示出优异的电化学性能。在电流密度为50 mA g−1时,前300 次循环的可逆容量为261 mAh g−1,库仑效率大于97%。在电流密度增大到100 mA g−1时,在1 000 次循环后可逆容量保持在211 mAh g−1,库伦效率仍高达98.2%。研究人员结合DFT 计算提出了Na+-GDY 配合物的可能构型(图10h),作者认为在充电过程中GDY 的Na+储存机制可能是一个混合作用过程,少部分Na+吸附在GDY 的表面和边缘(A2 和B1),大部分Na+嵌入在GDY 的大三角孔中。GDY 独特的微孔和中孔结构能够提供大量的Na+储存位点,且方便电子和离子的快速传输。孙靖宇等[105]通过理论计算发现GDY 理论储钾性能(620 mAh g−1)远高于石墨(278 mAh g−1),以此为依据对GDY 负极钾离子电池开展了实验研究。首先使用前人报道的Cu 片催化交叉偶联反应制备GDY,再经Ar 保护退火处理,去除了GDY 表面上的低聚物,提升活性材料的完整性和导电性。研究证明,热处理温度影响GDY 的结构和电化学性能。800 °C 煅烧处理后的样品G D Y-8 0 0,层间距从原来的0.37 nm 增大到0.41 nm,更加易于K+的穿梭和缓冲体积膨胀。以GDY-800 为负极的钾离子电池具有高的比容量(505 mAh g−1,电流50 mA g−1)、优异的速率性能(150 mAh g−1,电流5 000 mA g−1)和良好的循环稳定性(电流1 000 mA g−1下2 000次循环后容量效率超过90%)。总之,GDY 用做离子电池负极材料时优异的电化学性能主要归因于两个方面:一方面,GDY 中独特的sp 杂化碳为碱金属离子提供了丰富且强有力的吸附活性位点,增强了离子存储能力。另一方面,GDY 均匀分布的三角形孔洞结构有利于离子通过层间间隔的传输,同时也促进了离子在层间穿梭交互,从而提高循环稳定性和充放电速率。
以金属锌为阳极的水系二次锌离子电池(ZIBs)因其良好的安全性、低成本和理想的环境兼容性,在大规模储能和可穿戴柔性设备方面表现出巨大潜力。然而,枝晶问题和锌阳极的副反应(如腐蚀和析氢)等仍然严重影响ZIBs 电池的性能和循环使用寿命。研究发现在制备过程中引入GDY,可能有效解决上述问题。刘辉彪等[106]通过对GDY 进行氧化处理得到表面富含亲水性含氧官能团的氧化石墨炔(GDYO),并将其负载在锌片表面制备Zn-GDYO 复合阳极。研究发现GDYO 多孔结构、良好的亲水性及对Zn2+强的配位作用显著提升了阳极的性能。在充电过程GDYO 保证了电解液中Zn2+的均速穿梭并在阳极基片上的均匀还原沉积。和裸锌阳极相比,Zn-GDYO//Zn-GDYO 对称全电池的过电势降低、无锌枝晶生成,运行寿命增加了10 倍。这种Zn-GDYO 阳极还支持水系Zn-GDYO//ZnVO全电池的运行,在1 200 次循环后仍保持高的电池容量(94.5%)和库伦效率(99%)。另外,他们在亲水性聚四氟乙烯(PTFE)上通过真空过滤制备了GDYO 膜[107],作为Zn-MnO2电池的隔膜材料,同样可以提高电池比容量、倍率性能及循环稳定性。亲水性的多孔GDYO 隔膜,不仅能确保Zn2+的稳定传输,还促进质子介导的MnO2还原反应。同时,它可以保护MnO2阴极表面免受化学腐蚀,保持其完整性,有利于电子转移。近期,Yang 等[108]进一步揭示了N 掺杂GDY(NGDY)作为ZIBs 电池隔膜时,能够抑制锌枝晶生长的本质原因。NGDY 加速了水合Zn2+的去溶剂化,N 原子作为受体从Zn2+结合水分子中捕获电子,削弱了Zn2+与水分子的配位,促进了水分子的脱除。因此,电子可以从导电基底直接转移到Zn2+上,避免了O―H 键的减弱,稳定了界面电解液及电极界面处的pH,从而有效抑制锌枝晶的形成和碱式硫酸锌结晶的生成。研究证实,装配NGDY 隔膜的Zn//Zn 对称电池寿命提高了116 倍,Zn//V6O13电池在20.65 mA cm−2的超高电流密度和1.07 mAh cm−2的面容量下实现2 000 次循环。总之,GDY 对ZIBs 性能有很好地提升作用,为解决二次离子电池枝晶问题提供了一种新方法。
4.2.2 超级电容器
超级电容器是另一种重要的电能存储器件。炭材料也是其最常用的电极材料。GDY 有序的孔洞结构、宽的层间距以及大的比表面积,均有利于离子和电子的迁移和扩散,适宜于快速充放电,因此,GDY 在超级电容器中的电化学性能也被初步研究。Wang 等[109]发明了一种温和条件下在任意基材(如泡沫镍、泡沫铜、铜片、硅片等)上超快速生长超细GDY 纳米链的方法。即载有HEB 的有机液滴的基体放置于已预热板上,受热后迅速发生交联反应并膨胀,从而直接得到负载型的GDY 电极。所制备的泡沫镍基GDY 可直接用做无粘合剂超级电容器电极,所组装超级电容器的比面积电容等于53.66 mF cm−2,且循环性能稳定(1 300 次循环电容保持99%)。
Yue 等[110]为了研究N 掺杂对GDY 超电性能的影响,设计合成了含有42 个碳大环结构的氮掺杂三嗪基石墨二炔(TGDY)和相应未掺氮的含氢石墨二炔(HGDY)。对其超电性能研究发现,在电流密度为0.1 A g−1时,最优样品TGDY-800(800 °C 热处理后的TGDY)的比电容250 F g−1,高于HGDY-800 的比电容230 F g−1。TGDY 呈现二维层状堆叠结构和高的比表面积785 m2g−1,氮掺杂削弱了π-π 堆叠作用,致使其层间距(~0.45 nm)大于传统石墨烯(0.23 nm)和传统GDY 薄膜,这有利于电解质离子的穿梭扩散。N 掺杂能够增强GDY 的电子输运能力和离子扩散速率,高温热处理能够进一步增加材料的导电性并以提升其储能性能。
4.2.3 太阳能电池
GDY 作为一种空穴传输的大π 共轭材料,王吉政等[111]将其用作胶体量子点太阳能电池中的阳极缓冲层(图11a,b),发现GDY 能有效降低PbS 量子点的功函数,显著增强空穴从量子点活性层向阳极传输。含有GDY 阳极缓冲层的PbS 胶体量子点太阳能电池示意图(图11)。为了评估GDY 的作用,采用了不含GDY 层的太阳能电池作为对照组,该电池的性能如图11c,d 所示。含GDY 阳极缓冲层的器件的短路电流为22.83 mA cm−2,开路电压为0.654 V,填充因子等于72.14%;而对照组器件的Jsc为21.74 mA cm−2,VOC为0.650 V,填充因子等于67.34%。增加GDY 阳极缓冲层可将能量转换效率从9.49%提高到1 0.6 4%,光照下的有效载流子寿命从1.9 s 延长到2.4 s,表明器件内部载流子的复合几率减小,光生载流子有更长的时间移动,最终被金阳极收集。更长的载流子寿命和更小的复合阻抗均归因于GDY 中间层强化了PbS-EDT 层和Au 阳极的接触。另外,含有GDY 层的太阳能电池表现出优异的稳定性,在环境空气中放置120 天,各项性能指标衰减小于5%。
酒同钢等[112]成功合成石墨炔-氧化锌(GDZO)复合材料并将其应用于聚合物太阳能电池的电子传输层。研究发现,Zn 原子和O 原子均能和GDY 配位成键,形成C―Z n 键和C―O 键,这有利于ZnO 在GDY 上的均匀分散,形成高质量的中间层。与纯ZnO 薄膜(10%)相比,GDZO 太阳能电池能量转换效率提高至11.2%。GDY 的引入不仅加速了电子转移,而且减小了界面上电荷复合几率。同时,在90%的高湿环境下,GDZO 器件依然表现出很好的稳定性。GDZO 的态密度计算结果如图11e-g 所示,由于强的轨道杂化作用和GDY 与ZnO 之间新键的形成,在体相ZnO 的禁带之间的费米能级附近出现新的杂质态密度能级。分态密度结果表明杂质能级主要来自GDY 的碳原子。与体相ZnO相比,GDZO 中从最高占据孤立杂质能级和导带之间的能级差更小,所需电子激发能更小,电子更加容易激发。因此,GDZO 太阳能电池具有更好的载流子传输特性。
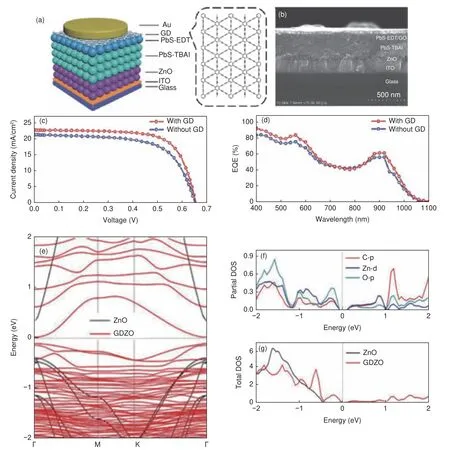
图11 (a) GD 阳极缓冲层的PbS CQD 太阳能电池示意图;(b)该电池的SEM 截面图;(c) 模拟AM 1.5 G 辐照的J-V 特性曲线和(d) EQE 光谱[111];(e)ZnO 和GDZO 的能带结构;ZnO 和GDZO 的(f)总态密度和(g)部分态密度[112]Fig.11 (a) Schematic diagram of PbS CQD solar cell with GD anode buffer layer; (b) SEM cross-section of the battery;(c) Simulated J-V characteristic curve and (d) EQE spectrum under irradiation[111]; (e) Band structure of ZnO and GDZO ;(f) Total state density and (g)partial state density of ZnO and GDZO[112].Reprinted with permission
在太阳能电池中引入GDY,不但能够有效地提高电池能量转化效率,而且改善器件的使用稳定性,若能大规模生产高性能功能化GDY,这可能为推动其实现商业应用提供重要支撑。
4.2.4 电催化水分解
析氧反应和析氢反应是水分解的两个半反应,开发高效稳定的催化剂一直是电催化水分解领域的关键。
薛玉瑞等[113]以泡沫镍为支撑的铜镍双金属氢氧化物纳米片基底,而后通过原位生长GDY,得到一系列水滑石型(LDH) 电催化剂NixCu1–xLDH@GDY/NF(0≤x≤1)。在O2饱和的1.0 mol L−1KOH 电解液中进行OER 性能测试发现,Ni0.74Cu0.26LDH@GDY/NF 具有最好的析氧活性,在10 mA cm−2电流密度下,过电位仅为292 mV,显著优于单金属水滑石型NiLDH@GDY/NF(414 mV)和CuLDH@GDY/NF(367 mV)及未复合GDY 的Ni0.74Cu0.26LDH/NF(356 mV)及其他C u/N i 比例对照组电催化剂。同时,Ni0.74Cu0.26LDH@GDY/NF 的Tafel 斜率(103 mV dec−1)小于NiLDH@GDY/NF(115 mV dec−1)和Ni0.74Cu0.26LDH/NF(147 mV dec−1), 证明前者OER 反应动力学过程更快。另外,目标催化剂6 000 圈连续的循环测试后OER 活性几乎无衰减。GDY 的引入有效增加铜镍水滑石的电化学活性面积和催化活性位点,电导率增加促进了电荷转移,而且能够保护催化剂不被腐蚀,从而提高复合催化剂的整体性能。
卢秀利等[114]合成了镍硼氧化物/石墨炔(NiBi/GDY)用于光/电催化OER 研究。以伊红Y(EY)为光敏剂,三乙醇胺(TEOA)为牺牲剂,NiBi/GDY 在可见光照射下(>400 nm)的OER 速率为4.54 mmol g−1h−1,比NiBi/GO 和NiBi高2.9 倍和4.5 倍。由于NiBi与GDY 强耦合作用,促进了EY 中光生电子和空穴的分离,NiBi/GDY表现出极高的光催化HER 活性和稳定性。同时,NiBi/GDY 在1.0 mol L−1KOH 溶液中也表现出了较好的电催化HER 活性,过电位478.0 mV 时的电流密度为400 mA/cm2,优于商用Pt/C(505.3 mV@400 mA/cm2)。研究者认为,NiBi/GDY 催化活性的提升主要与GDY 特殊π 共轭结构、强的给电子效应及高的电化学活性面积有关。
高电流密度(≥1 A cm−2)是衡量水分解尤其是海水分解电催化剂能否大规模工业应用的关键因素。然而,由于催化剂中缺乏稳定的催化活性位点,达到高电流密度仍然是一个巨大的挑战。郭彦炳小组[115]研制了一种三维自支撑石墨二炔/氧化钼(GDY/MoO3)材料,通过理性设计“sp C―O―Mo 杂化”界面来实现高效的HER。“sp C―O―Mo 杂化”创造了新的本征催化活性位点(非氧空位位点)同时大大增加了活性位点的数量(比单独MoO3高8 倍)。“sp C―O―Mo 杂化”促进电荷转移并促进水分子的解离过程,具有出色的HER 活性。在碱性电解液中HER 电流密度大于1.2 A cm−2。在天然海水中也表现出中较好的HER 活性和稳定性。
赵英杰等[116]以多炔基氟苯为前体在碳布上原位生长了柔性三维多孔氟代石墨炔电催化剂(p-FGDY/CC),并将其作水还原和氧化的双功能催化剂。p-FGDY/CC 具有超亲水表面和高柔韧性,非常适合作为水分解的工作电极,亲水表面可促进反应物和产物在电催化剂界面的传输和接触,从而有助于改善其催化性能。该材料在全pH 范围内均显示出优异的电催化性能,在1.0 mol L−1KOH 的HER 过程中,达到10 mA cm−2电流密度仅需82 mV(vs.RHE)过电位,优于其他非金属催化剂,甚至可以与金属基催化剂相媲美。即使在强酸性条件下(0.5 mol L−1H2SO4),p-FGDY/CC 仍表现出良好的HER 性能,具有较小的过电位(10 mA cm−1时为92 mV)。此外,还评估了p-FGDY/CC 在碱性和酸性电解质中的OER 性能,在1.0 mol L−1KOH 溶液中,P-FGDY/CC的起始电位为1.54 V(vs.RHE),达到10 mA cm−2所需电位为1.69 V,其在碱性条件下的活性远超大多数已报道的优质非金属催化剂,且稳定性突出。这归因于强的F―C 键合改变了局部p-p 电子轨道耦合相关的电子轨道填充,从而增强了C2 位点的富电子特性,明显提高了电子转移能力,确保了对各种O/H 中间体吸附/脱附更高的选择性。因此,p-FGDY/CC 在强酸和强碱电解液中对HER 和OER 均具有良好的催化性能。
4.2.5 电化学氧还原
电催化氧还原反应是燃料电池和金属-空气电池中的基本半反应,是近期电化学领域的又一个研究热点。因其较高的过电位,ORR 是一个缓慢的动力学过程,因此,亟待开发高效的催化剂以推动其市场化应用。传统的ORR 催化剂以贵金属Pt 为主,开发价格低廉的非贵金属催化剂是促进燃料及金属-空气电池规模应用的必然选择。GDY 通过掺杂、修饰、担载,可以构筑成为各种高效的ORR 催化剂。
如前文所述,王丹团队[60]通过周环反应成功引入一种新型掺杂N 原子,实现了GDY 炔键上sp―N 的定点、定量掺杂,并利用XANES 和XPS确定了sp―N 原子掺杂位点及原子含量(NFLGDY-900c,1.36%)。研究证明,该材料在碱性条件下,半波电位为0.87 V,在0.75 V 时电流密度38 mA cm−2,活性高于商业Pt/C 催化剂,并表现出更好的稳定性和甲醇耐受性;在酸性条件下,该材料的活性虽略低于商业Pt/C 催化剂,但仍远优于其它非金属催化剂。和吡啶N 相比,sp―N 对ORR 发挥更加重要的影响。DFT 计算发现,与其它形式的掺N 构型相比,sp―N 原子的引入使得其周围碳原子带有更多的正电荷,更有利于O2的吸附和活化,使电子更易转移到催化剂表面,从而显著提高ORR 催化性能。
蔡称心等[117]在NaBH4水溶液中通过还原吸附在GDY 表面的Fe3+离子合成了Fe-GDY 催化剂,并在0.1 mol L−1KOH 溶液中对其ORR 活性进行了评估。研究证明,GDY 是一种很好的单原子催化剂基底材料,可以通过形成Fe-C 共价键来锚定Fe 原子。Fe-GDY 上ORR 的阴极峰电位为53 mV,与Pt/C(20%)催化剂的阴极峰电位相当,相应的峰电流约为0.42 mA cm−2,是Pt/C 催化剂的1.68 倍。Fe-GDY 催化剂在抑制二电子转移反应的同时,促进了四电子ORR 反应的进行,表现出较高的催化选择性。
4.3 石墨炔在生物医学领域的应用
随着纳米生物技术的进步,纳米材料在癌症治疗应用方面获得长足的发展[118–119]。作为一种新型的二维炭材料,GDY 在抗菌及癌症治疗等方面的应用研究也越来越多。
细菌一直以来威胁着公众健康,抗生素一直是治疗细菌感染的首选药物。然而,在抗感染治疗过程中长期使用抗生素会导致耐药细菌不断出现,致使治疗效果下降。因此,新型非抗生素治疗材料应用而生。GDY 可作为非抗生素抑菌材料,研究发现,GDY 和GDYO 既可通过与细菌的直接接触杀菌,也可利用可见光照射产生活性氧物种杀菌[120]。GDY 与其他杀菌剂配合使用,可能实现更好的协同杀菌作用,如GDY 与银纳米颗粒(Ag NPs)协同可增强抗菌能力[121],GDY@Ag Nps 对枯草芽孢杆菌和大肠杆菌的最低抑菌浓度分别为0.24 和1.2 μg mL−1,杀菌性能比Ag NPs 和目前报道的GO@Ag NPs 更优异。GDY@Ag NPs 比报道的GO@Ag NPs (+20.3 mV)具有更高的正电荷密度,约为+38±0.2 mV,这增强了其与带负电荷细菌的相互作用。GDY 的抗菌能力被认为是物理作用与化学作用结合的结果,带正电的GDY 有利于用包裹在带负电荷的细菌表面,GDY 纳米片可以直接插入和破坏细菌细胞膜,从而阻止细菌生长。
基于纳米酶的抗菌治疗被认为是一种有前景的新技术,具有广谱抗菌效果。然而,目前纳米酶的催化活性较低,严重阻碍了其抗菌效果。为了开发更有效的抗菌纳米酶,利用协同效应提高酶活性,Bai 等[122]开发了一种基于GDY 纳米壁包裹的空心硫化铜纳米立方体(CuS@GDY)异质结复合材料,该材料在近红外区域具有强的局域表面等离子体共振响应。这种纳米酶可以捕获等离子体共振诱导的热载流子并产生光热效应,在808 nm 光照时类过氧化物酶活性显著增加。其对多种病原体(包括耐甲氧西林-金黄色葡萄球菌、金黄色葡萄球菌和大肠杆菌)均具有快速、高效、广谱的抗菌活性,这与它独特的分级结构、窄的GDY 纳米壁的带隙、CuS 纳米笼的LSPR 效应、快速的界面电子转移和CuS@GDY上的碳空位均密不可分。该课题组还开发了一种可用于抗感染治疗的生物兼容性复合材料AuAg 纳米笼/石墨二炔@聚乙二醇(AuAg/GDY@PEG)[123]。基于其宽的吸光能力和优良的光热转换能力,采用AuAg/GDY@PEG 可以对鼠伤寒沙门氏菌进行光热免疫分析。同时,AuAg/GDY@PEG还表现出了优异的广谱光热杀菌性能,对金黄色葡萄球菌和大肠杆菌的杀灭率为99.999%。
GDY 在癌症的多模式疗法中也具有潜在的应用前景。王树等[124]使用聚乙二醇对GDY 纳米片修饰(GDY-PEG)以提高水溶性和生物相容性,将GDY 首次成功地用作活体小鼠体内癌细胞光声成像和光热治疗的光-热-声波纳米换能器。由于GDY 在近红外区有较大的摩尔吸光系数,GDY-PEG 表现出优异的光声响应和光热性能,光热转换效率高达42%。刘辉彪等[125]开发了一种用于癌症光热/化疗组合治疗的GDY 纳米片药物递送系统。GDY 纳米片拥有突出的光热转换能力和载药效率,通过π-π 堆积和静电相互作用可以将模拟药物阿霉素(DOX)负载其上,GDY/DOX 药物递送系统在小鼠活体试验中显示出pH/光双重响应释放行为,近红外激光照射下(波长808 nm)药物可以高效释放。此外,组织病理学和血清生化分析表明,GDY 具有良好的生物相容性,且无明显副作用。细胞和动物模型实验均证明利用该体系进行光热/化疗组合治疗能够提高抗肿瘤效果,且生物安全性良好。GDY 还能够作为载体材料辅助芬顿反应的发生,通过铁离子将内源性过氧化氢催化转化为具有活性的·OH,有效实现由芬顿反应介导的肿瘤治疗。胡建设等[126]开发了一种基于石墨二炔氧化物(GDYO)的肿瘤靶向治疗的海绵铁纳米复合材料。其合成过程如图12a 所示,通过溶剂热法将Fe3O4纳米粒子沉积在GDYO 表面制取rGDYO-Fe3O4。为了赋予该材料更好的生物相容性和肿瘤靶向能力,选择肿瘤靶向聚合物DSPE-PEG2000-CREKA 对其进一步修饰,得到纳米复合物rGDYO-Fe3O4-CREKA,即TTIS。在CREKA 多肽和血纤蛋白的强亲和作用下,TTIS 可以朝着肿瘤位点定向转移并不断富集。TTIS 的近红外热成像照片(图12b)可以直观地观察到随着浓度和辐照时间增加,温度会随之升高,这表明TTIS 可以有效地将近红外辐照转化为热能。近红外照射下,TTIS 可以提高肿瘤部位的温度,达到有效的光热治疗效果,同时产生的热量可以加速酸性肿瘤环境下Fe2+/Fe3+离子的释放,并提高Fenton 反应速率以快速生成·OH(图12c),从而成功实现光热治疗和限域芬顿反应介导的联合癌症治疗。另外,GDYO 对于癌症的免疫治疗也有较好地效果。肿瘤相关巨噬细胞是一种重要的癌症免疫治疗细胞群。Chen[127]发现GDYO 可以极化M2 巨噬细胞经过活化促炎性路径来杀死癌细胞,并且荷瘤小鼠模型实验中,腹腔内注射GDYO可以抑制黑色素瘤的生长。此外,GDYO 通过巨噬细胞直接或间接活化细胞毒性T 细胞,在乳腺癌抗癌模型实验中增强免疫检查点抑制剂的应答。GDYO 能够激活免疫系统的先天和适应性两种机制,提高癌症免疫治疗的疗效。
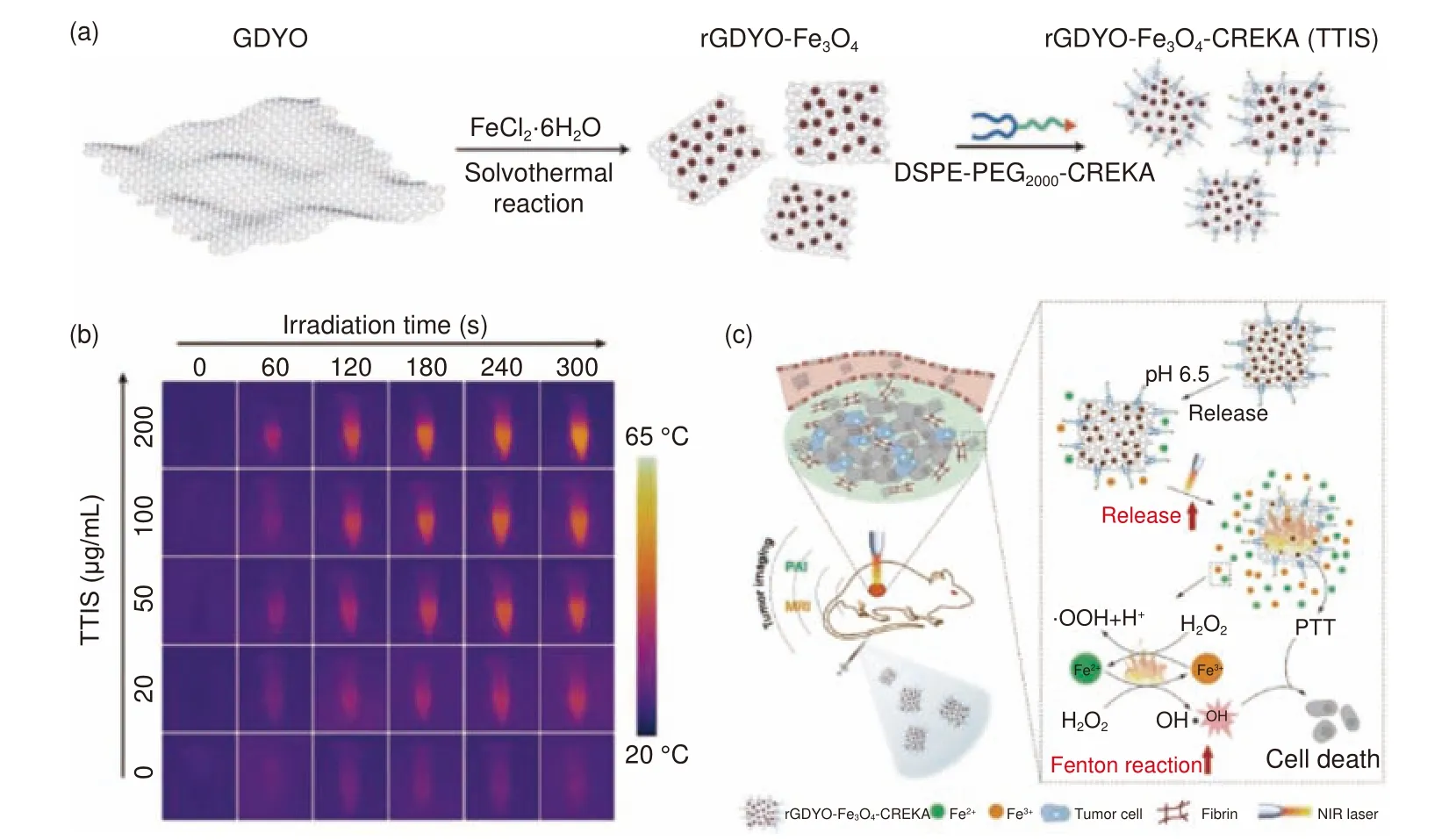
图12 (a) TTIS 制备示意图;(b) TTIS 在激光照射下的近红外照片;(c) TTIS 介导光热增强芬顿反应的肿瘤治疗示意图[126]Fig.12 (a) Schematic illustration of the fabrication of TTIS; (b) corresponding near-infrared photographs of TTIS under laser irradiation; (c) Schematic illustration of TTIS mediated tumor therapy via a photothermally enhanced Fenton reaction[126].Reprinted with permission
放射疗法在肿瘤治疗中的作用和地位日益突出,已成为治疗恶性肿瘤的主要手段之一。高能电离辐射能强有力地破坏癌细胞,但同时也会不可避免地损害周围的正常细胞。使用GDY 纳米粒子制成辐射保护剂能够保护正常组织免受辐射引起的损伤。谷战军等[128]合成了牛血清白蛋白修饰石墨二炔纳米粒子(GDY-BAS NPs),并研究其自由基捕获能力和细胞和动物体组织的辐射损伤保护性能。自由基淬灭实验表明,GDY-BAS NPs 具有较强的自由基淬灭能力,能有效清除1,1-二苯基-2-苦基肼自由基、2 ,2-联氮-二(3-乙基-苯并噻唑-6-磺酸)自由基、·O2−和·OH 等自由基。体外细胞实验证明,GDY-BAS NPs 有效消除细胞内活性氧物种,减轻辐射对其DNA 的损伤,显著提高细胞在电离辐射下的生存能力。体内动物实验表明,GDY-BAS NPs 能保护小鼠骨髓DNA 免受辐射损伤,并使肝、肺等主要器官的超氧化物歧化酶和丙二醛恢复到正常水平。同时,生物安全分析结果证明,该材料在小鼠体内呈现良好的生物相容性和可忽略的体内毒性。该研究为放疗过程中降低基体组织辐照损伤水平提供了一种全新的防护材料,为GDY 在生物医学领域的应用开辟了一条新的途径。
5 结论与展望
综上所述,GDY 是一类sp 和sp2杂化碳原子共存的碳同素异形体,具有大π 共轭结构和尺寸可调的碳多孔骨架。自2010 年首次合成后,GDY 因其独特结构和特异的物理化学性能,一直吸引着研究者们的探索兴趣。本文重点对近年来GDY 在制备方法、修饰改性及典型应用的进展进行了综述。制备方法部分主要介绍了李玉良院士原创的铜箔为基底和催化剂的Glaser-Hay 交叉偶联法,以及其它典型的合成方法,包括化学气相沉积法、范德华外延法、微波诱导法、爆炸法和双极电化学法等。其次,归纳了理论计算和实验方面有关非金属、金属掺杂改性GDY的性能变化和调控机理。最后,重点阐述了GDY 基材料在环境、能源和生物医学等领域的典型应用,证明GDY 类二维材料具有光明的发展前景。
尽管GDY 合成方面已取得了一系列重要的研究成果,但其研究仍处于起步阶段,挑战和机遇依然存在。GDY 的未来发展仍然面临众多难题及困难需要解决:第一,开发工艺简单、条件温和、成本低廉、且适合大规模生产GDY 的合成方法,从而进一步挖掘其在不同领域应用潜力。特别是迫切需要建立不同炔键数目、不同晶体尺寸和单层/少层的高质量GDY 的可控合成策略,从而分别拓展其基础和应用研究的深度和广度。第二,GDY 作为二维材料是一种优良的载体材料,但要改善其在电化学性能,其电导率尚需提高。如何通过自下而上的分子合成实现非金属杂原子在固定位点上的精准掺杂,并明确掺杂位点和掺杂量对改性GDY 物化性能的调控机制,是GDY 改性的重点。第三,虽然在GDY 功能化的理论计算方面取得了一定的进展,但在实验验证和实际应用方面仍处于初级阶段。单原子催化已成为当前催化科学的热点,利用金属原子与GDY 之间强的配位作用,合成高分散、高负载量、高活性的单原子催化剂,并努力寻找相匹配高附加值的医药合成、化工生产的催化反应,具有重要的现实意义。第四,将理论计算模拟和原位分析表征手段相结合,从分子水平甚至原子水平深入理解GDY 基材料结构、性能和应用之间的本质联系,从而更加全面、更加深入地研究和利用GDY。例如,通过大尺度分子动力学模拟,探索GDY 在癌症治疗过程中与生物活体内的生物大分子之间的作用机理;通过理论计算和原位分析的相互佐证,全面揭示GDY 基催化剂中的催化活性位点等。在无数研究人员的共同努力下,GDY 的发展将会为碳质材料的明天带来更大的机遇,终将在众多领域开花结果。
致谢
感谢国家自然科学基金项目(22276144,22076151);陕西省自然科学基础研究计划项目(2020JM-562);陕西省教育厅重点科学研究计划项目(20JS054)的支持。
猜你喜欢
杂志排行

新型炭材料的其它文章
- Coal-based graphene as a promoter of TiO2 catalytic activity for the photocatalytic degradation of organic dyes
- 咖啡渣成型制备生物质炭及其CH4/N2分离性能
- A flexible hard carbon microsphere/MXene film as a high-performance anode for sodium-ion storage
- Preparation and lithium storage of anthracite-based graphite anode materials
- Oxygen-incorporated carbon nitride porous nanosheets for highly efficient photoelectrocatalytic CO2 reduction to formate
- 炭纸衬底上化学气相沉积直立型二维过渡金属硫化物及其电催化产氢性能