炭纸衬底上化学气相沉积直立型二维过渡金属硫化物及其电催化产氢性能
2022-12-13姚孝璋HitanshuKumar
王 克,汤 飞,姚孝璋,Hitanshu Kumar,2,干 林,*
(1.清华大学深圳国际研究生院,材料研究院,广东 深圳 518055;2.深圳大学,材料学院,广东 深圳 518055)
1 前言
氢能来源清洁且具有循环可持续性,被认为是最有可能替代传统化石燃料的能源[1,2]。氢能的来源相对广泛,包括水分解、循环热过程以及生物质过程等,其中,来自(光)电解水的析氢反应(HER)是为氢能相关技术如燃料电池提供高纯度的氢燃料的有吸引力的途径[3–6]。虽然Pt 基催化剂是纯金属中催化活性最高的HER 催化剂,但其高额的成本和有限的储量阻碍了其大规模生产和应用[7]。近年来,二维过渡金属硫化物(TMDs)如MoS2、WS2等,因其相对较低的成本和较好的产氢催化活性,作为非贵金属HER 电催化剂引起了广泛的关注[8,9]。人们先后开发了多种制备二维过渡金属硫化物的方法,包括分子束外延法[10,11]、化学气相沉积[12,13]、有机金属化学气相沉积[14]、水热还原法[15]等,其中,化学气相沉积法因可以在不同衬底上合成高质量的二维过渡金属硫化物,尤为值得关注[16,17]。
目前,限制二维过渡金属硫化物的产氢催化性能主要由两个方面的因素。一是由于其半导体的特性,导电性相对金属催化剂较差,在一定程度上限制了电催化过程中的电子传递过程[18]。并且,采用传统的粉末性过渡金属硫化物制作电催化电极时还常需要用黏结剂,进一步限制了其导电能力。炭纸是经济易得的材料,具有孔隙多、导电性好、耐酸碱的优点,已被广泛用于电化学研究领域,尤其电催化载体、超级电容器等,可改善导电性较差的材料的电子传导,充分发挥电极材料的电化学性能。同时,炭纤维与材料连接较为牢固,避免了使用黏结剂对精细电化学测试的影响[19,20]。但以炭纸为基底化学气相沉积不同的二维过渡金属硫化物还鲜有系统的研究,目前仅有文献报道过炭纸表面生长MoS2纳米片[21]。二是过渡金属硫化物的产氢活性位很有限。譬如,研究证明MoS2中相对稀缺的边缘Mo 位是HER 的活性位点,而平面S 是惰性的[22,23]。近来,研究发现通过在平面内引入硫(S)空位是提高HER 催化活性的有效途径[24],尤其是Tsai 等报道,通过简单的电化学还原法使MoS2中的S2-与原位产生的H2结合生成气态的H2S,从而产生平面内大量的单个原子级S 空位,以提高HER 活性[25]。但这种电化学阴极活化引入S 空位的方法是否适用于其他过渡金属硫化物以及S 空位的形貌和结构还有待研究。
因此,作者系统研究了采用了化学气相沉积方法直接在炭纸上生长了3 种代表性的二维TMDs(MoS2,NbS2,WS2)的制备工艺,并以其作为无黏结剂的一体化产氢催化电极。通过优化生长工艺,成功制备出了3 种直立型二维过渡金属硫化物,结合扫描电子显微镜和透射电子显微镜及X-射线衍射对不同合成条件下制备的产物的微观形貌和结构进行了表征,并系统比较研究了其HER 电催化性能。为进一步提高其HER 催化活性,采用电化学还原法在这3 种过渡金属硫化物中引入S 缺陷,并结合透射电子显微镜和原位电化学拉曼光谱方法研究了处理前后的结构差异。这些结果对优化炭纸基底上化学气相沉积不同过渡金属硫化物的合成条件以及理解其结构与产氢性能之间的联系具有一定的指导意义。
2 实验
2.1 试剂
五氯化钼(MoCl5,纯度:≥99.5%)、五氯化铌(NbCl5,纯度:≥99.5%)、六氯化钨(WCl6,纯度:≥99%)购于Alfa-Aesar 公司,硫粉(S,纯度:≥99%)购于阿拉丁,浓硫酸(质量分数≥98%)购于国药,炭纸(Toray 060)上海河森公司。
2.2 实验过程
2.2.1 化学气相沉积合成MoS2、NbS2和WS2
常压化学气相沉积(CVD),反应器为高温管式炉炉,石英管管径为70 mm。如图1 所示,先将石英管中通入Ar,保持惰性气氛,然后迅速(30 s之内)将前驱体MoCl5和硫粉分别放置于炉管的不同区域,使用加热带加热至~200 和~250 °C,用量分别为~0.2 和~0.4 g。将炭纸放置于炉膛的α 区,以10 °C/min 的升温速率,使反应区中心温度到达设定值(750 或850 °C)。反应中通入4% H2/Ar(12 L/h),反应时间30 min,最后在Ar 气氛中冷却至室温,获得生长在衬底上的MoS2[26]。
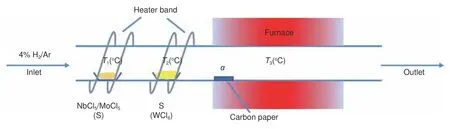
图1 TMDs(MoS2、NbS2、WS2)的合成示意图Fig.1 Schematic of synthesis of TMDs (MoS2, NbS2, WS2)
生长NbS2时,前驱体NbCl5和硫粉的用量分别为~0.2 和~0.5 g。设定反应区中心温度800 °C,反应中通入4% H2/Ar(12 或40 L/h),生长时间30 min。反应结束后,在S 和Ar 的气氛中冷却至350 °C,最后在Ar 的气氛中冷却至室温[27]。其余条件与生长MoS2类似。
生长WS2的不同之处在于,前驱体硫粉(~0.2 g)和WCl6(~0.4 g)分别放置于炉管的不同区域,使用加热带加热至~250 和~300 °C。反应中通入4% H2/Ar(12 或40 L/h),设定反应区中心温度(750 或850 °C),生长时间30 min,最后随炉冷却[28]。
2.2.2 材料表征
采用扫描电子显微镜(SEM,ZEISS Supra 55-VP),配置有牛津仪器公司的X-MAX 80T EDS(Energy dispersive X-ray spectrum, EDX)探头获得不同衬底上生长的3 种过渡金属硫化物的微观形貌图以及原始样品的元素成分比。透射电子显微镜(TEM),低倍的显微像和大区域的元素成分分布在美国FEI 公司Tecnai G2Spirit T12(120 kV)上进行,配置有牛津仪器公司的X-MAX 80T EDS(Energy dispersive X-ray spectrum, EDX)探头。高分辨透射电子显微(HRTEM)像在Tecnai G2F30 场发射透射电镜(300 kV)上采集。
电化学测试:将生长有过渡金属硫化物的炭纸裁剪为直径约2.5 mm 的圆片,用镊子借助水的浸润性紧贴在直径为5 mm 的玻碳电极并使其位于玻璃炭的中心,套上定制的中心孔直径2 mm的塑料盖帽,压紧使盖帽、炭纸、玻璃炭基底三者贴合,放入50 °C 的烘箱中烘20 min 后作为工作电极。HER 电催化活性采用电化学工作站(Biologic SP200)在三室电化学池中进行,其中工作电极加载在旋转圆盘电极装置上(美国Pine Instrument),参比电极为Hg/Hg2SO4电极(经校准后相对可逆氢电极的电势0.720 V),对电极为石墨棒,电解液采用0.05 mol/L H2SO4。HER 均在H2饱和的条件下测试,扫速为5 mV/s,转速为1 600 r/min,测试过程中均采用IR 电阻矫正。为了提高过渡金属硫化物的催化活性,将其在−1 V(相对于可逆氢电极(vs.RHE))下恒电位保持3 min,以实现电化学还原活化形成S 空位缺陷。
原位电化学-拉曼光谱(in situ electrochemical Raman spectroscopy)采用配备532 nm 激光的拉曼光谱仪(Horiba Lab RAM HR800)在定制的原位拉曼电化学池中进行,以铂丝和Ag/AgCl 为对电极和参比电极,将CVD 法合成的带有样品的直径为4 mm 的圆形炭纸放在直径为3 mm 的玻璃碳上作为工作电极,电解液为0.05 mol/L 硫酸溶液。采用计时电流法(Chronoamperometry,CA)测试,在不同恒电位下(0~1.4 V)保持2 min 后收集催化剂的拉曼光谱,4 次采集,每次10 s,直至40 s 的采集结束,获得一个图谱数据。
3 结果与讨论
3.1 炭纸表面化学气相沉积MoS2、NbS2 和WS2 的合成条件
采用管式炉,MoCl5和硫粉作为前驱体,在4% H2/Ar 气氛下,用CVD 的方法制备了二维的MoS2。控制4% H2/Ar 的流速为12 L/h 不变,研究了合成温度对MoS2形貌的影响。图2(a-b)给出了750 °C 下生长的MoS2的SEM 照片,显示炭纤维表面出现了大量直立二维片状结构,单片的平面直径大概500 nm。在850 °C 的下生长的MoS2(图2(c-d))相对稀疏,但仍呈直立片状结构,单片的直径也接近500 nm。EDX 分析表明750 °C 时S∶Mo=2.6,而850 °C 是S∶Mo=2,表明较低温度下S 元素偏多。接着对成分比为2 的MoS2进行TEM 分析,形貌照片(图2e)表明了其与前述S E M 类似的片层结构,高分辨照片(图2f)表明了其良好的结晶性,相关的晶面及晶面间距标注在图片及其快速傅里叶变换(FFT)图中,与2H 相的MoS2相对应。尺寸分布柱状图(图2g)表明其片的大小均匀,为500 nm 左右。对XRD 测试结果(图2h)表明,两种温度下生长的MoS2均是2H 相。遗憾的是低温下有S 单质的峰出现作为杂质掺入到了MoS2中。这可能是因为温度较低,S 蒸气不能与H2较好地反应生成H2S,最终导致了S 单质的沉积。该结果表明,反应温度对二维过渡金属硫化物的成分有重要影响,在850 °C 下能合成更纯的MoS2。
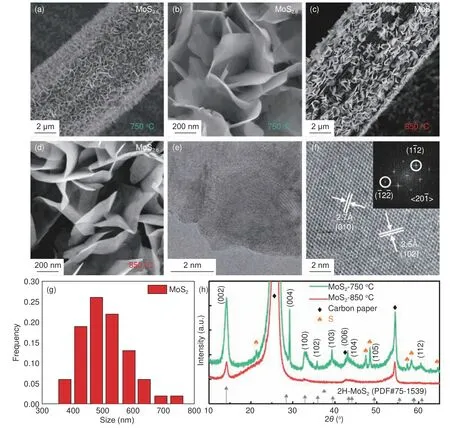
图2 在12 L/h 的气体流速下,生长在碳纸上的MoSx 的SEM 照片:750 °C 的(a)低倍和(b)高倍照片,850 °C 的(c)低倍和(d)高倍照片;12 L/h 的气体流速下850 °C 生长的MoS2 的(e)TEM 照片和(f)HRTEM 照片,内嵌图为其FFT 以及(g)尺寸分布图;(h)12 L/h 的气体流速下,750 °C 和850 °C 生长在碳纸上的MoSx 的XRD 谱图Fig.2 SEM images of synthesized MoSx on carbon paper with a flow of 12 L/h.(a-b) 750 °C, (c-d) 850 °C, (e) TEM image, (f) HRTEM image with FFT and(g) size distribution of MoS2 on carbon paper with a flow of 12 L/h under 850 °C and (h) XRD patterns of synthesized MoSx on carbon paper with a flow of 12 L/h under 750 °C or 850 °C
进一步研究了炭纸表面化学气相沉积NbS2的制备工艺,尤其是反应气体的流量对制备工艺的影响。800 °C 下,在12 L/h H2/Ar 的气体流量下成功合成了直立型NbS2纳米片(图3(a-b)),单片的平面尺寸在500 nm~1 μm,但其厚度较大,不是很均匀。然而,增大气体流量到40 L/h 时,生长的NbS2(图5(c-d))单个纳米片的平面尺寸增大而厚度明显较小。进一步的EDX 分析表明在两种气体流量下生长的S∶Nb 的比值都接近2。40 L/h 时生长的NbS2的TEM 照片(图3g)能更清楚地看清其二维片层结构,HRTEM 照片(图3f)表明了良好的结晶性,晶面和晶面间距与3R 相的NbS2相对应。尺寸分布柱状图(图3g)表明其片的大小均匀,直径为1 μm 左右。而XRD 结果(图3h)也表明两种气体流速下生长出的都是3R 相的NbS2,这表明气体流量对二维过渡金属硫化物的成分与结构没有明显的影响。但在40 L/h 下,NbS2衍射峰强度更高,表明在相对大的气流下生长的NbS2的含量较高。
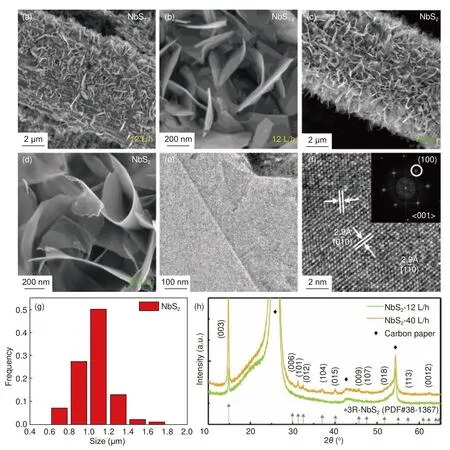
图3 在800 °C 下,生长在碳纸上的NbSx 的SEM 照片:12 L/h 的气体流速的(a)低倍和(b)高倍照片,40 L/h 的气体流速的(c)低倍和(d)高倍照片;在800 °C,12 L/h 的气体流速下生长的NbS2 的(e)TEM 照片和(f)HRTEM 照片,内嵌图为其FFT 以及(g)尺寸分布图;(h)在800 °C,12 L/h 和40 L/h 的气体流速下,生长在碳纸上的NbS2 的XRD 图谱Fig.3 SEM images of synthesized NbS2 on carbon paper under 750 °C with a flow of (a-b) 12 L/h, (c-d) 40 L/h, (e) TEM image, (f) HRTEM image with FFT and (g) size distribution of NbS2 on carbon paper under 800 °C with a flow of 40 L/h and (h) XRD patterns of synthesized NbS2 on carbon paper under 800 °C with a flow of 12 L/h or 40 L/h
采用类似的CVD 方法,以S 和WCl6作为前驱体,继续以炭纸作为基底研究了WS2的合成以及载气流量和反应温度的影响。与NbS2的合成结果类似,在固定温度850 °C 的条件下,当载气流量从4 L/h 提高到12 L/h 时(图4(a-d)),所生长的‘WS2’在炭纤维上的密度显著增加,但EDX 分析发现,S 与W 的原子比(3.2~2.8)均明显大于理论比值2,说明合成的材料并不全是WS2。当保持4% H2/Ar 的气体流速为12 L/h 不变,将反应温度从850 °C 降至750 °C(图4(e-f)),得到S∶W 的值从2.8 减小到了1.9,更加接近理想的原子比值2。与前述MoS2的结果一致,这再次验证了反应温度对过渡金属硫化物的温度有显著影响。有意思的是,此时生长的WS2呈现出一种特殊的纳米纤维/纳米片的多级结构(图4g),其中平面尺寸大约为100~200 nm(图4i),WS2纳米片沿一维方向自组装成一维纳米结构。HRTEM照片(图4h)标注的晶面及晶面间距与2H 相的WS2对应。进一步的XRD 分析(图4j)表明,750 °C,12 L/h 流量下生长得到的是2H 相的WS2。
图4 三种合成的WSx 的SEM 图:(a-b)850 °C 及4 L/h 的气体流速,(c-d)850 °C 及12 L/h 的气体流速,(e-f)750 °C 及12 L/h 的气体流速;在750 °C,12 L/h 的气体流速下生长的WS2 的(g)TEM 照片和(h)HRTEM 照片,内嵌图为其FFT 以及(i)尺寸分布图;(j)在750 °C,12 L/h 的气体流速下,生长在碳纸上的WS2 的XRD 图谱Fig.4 SEM images of three types of synthesized WSx on carbon paper (a-b) under 850 °C with a flow of 4 L/h, (c-d) under 850 °C with a flow of 12 L/h,(e-f) under 750 °C with a flow of 12 L/h.(g) TEM image, (h) HRTEM image with FFT, (i) size distribution and (j) XRD patterns of WS2 on carbon paper under 750 °C with a flow of 12 L/h
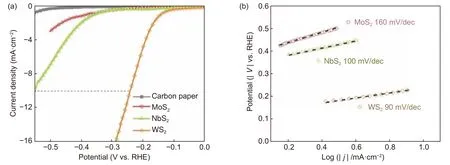
图5 三种TMDs 在0.05 mol L−1 H2SO4 中的HER 电催化性能:(a)碳纸及MoS2 (850 °C, 12 L/h)、NbS2 (800 °C, 40 L/h)、WS2 (750 °C, 12 L/h)的HER 极化曲线,(b)MoS2、NbS2、WS2 的Tafel 斜率图Fig.5 (a) HER plots and (b) Tafel slopes of MoS2, NbS2 and WS2
对比合成的3 种成分最接近S∶M(M=Mo、Nb、W)值为2 的TMDs,发现其尺寸的大小是由反应物的浓度(气体流速影响)和生长速率(温度影响)以及反应的难易程度共同决定的。在气流足够的情况下,生长速率越小生长的样品尺寸也越小[17]。WS2的合成温度750 °C,在三者中温度最低,生长速率最小,致使生长的单片尺寸最小。
3.2 不同二维过渡金属硫化物纳米片/炭纸电极的产氢催化性能
在上述结果基础上,挑选了3 种最佳合成条件下得到的成分接近MS2(M=Mo、Nb、W)的TMDs,由于其与炭纸结合紧密,导电性得到改善,直接作为电极进行HER 性能测试。结果表明,3 种TMDs 的HER 催化性能依次为WS2>MoS2>NbS2,其中WS2和NbS2在10 mA/cm2的过电位分别为250 mV 和550 mV。同时,WS2拥有最小的Tafel 斜率90 mV/dec;NbS2的Tafel 斜率次之,为100 mV/dec;而MoS2的Tafel 斜率最大,达到160 mV/dec。这些结果均表明WS2的HER性能最佳。虽然之前的报道发现WS2边缘位点的理论活性比MoS2的边缘位的活性略低[29],但SEM 照片中可以看出WS2中暴露的边缘位更多,因此活性最好[30]。
3.3 二维过渡金属硫化物的阴极电化学活化
根据之前的报道,电化学阴极还原活化可以明显提高MoS2的电催化HER 性能。其具体原理是在−1 V(vs.RHE)下保持3 min,使MoS2中的S2−与原位产生的H2结合生成气态的H2S,从而产生平面的S 空位,以提高HER 活性[25]。采用同样的方法对CVD 方法合成的MoS2和NbS2分别进行了电化学阴极还原活化,发现它们对HER 的催化性能均得到明显提高。其中,在−0.45 V(vs.RHE)的电位下,MoS2的产氢电流密度增大到大约原来的3 倍(图6a);而NbS2在−0.45 V 的产氢性能也提高了50%;对于WS2也有小幅度的提升。证明了此种阴极活化方式是一种提高TMDs的HER 活性的普适性方法。
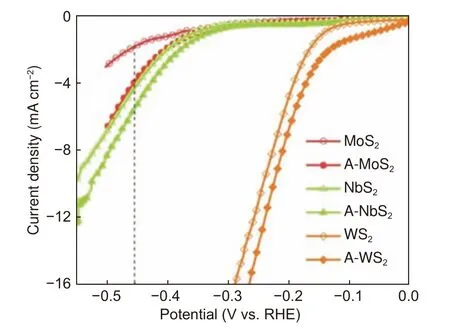
图6 MoS2、NbS2 和WS2 在0.05 mol L−1 H2SO4 中阴极电化学活化(-1.0 V, 3 min)前后的HER 极化曲线Fig.6 HER plots of MoS2, NbS2 and WS2 before and after electrochemical cathodic activation
活化后的MoS2(图7a)中出现了较多晶格畸变(如倾斜程度不同的白色虚线所示),此区域的成分分析证明S∶Mo 的值为1.8,小于原始样中的比值2,表明S 空位缺陷的产生。并且整个区域无明显的大面积S 空位区,表明S 空位的大小在原子级。这种电化学阴极还原活化使MoS2表面出现了大量的单个S 空位缺陷,可显著提高其产氢性能[25]。不同的是,NbS2在电化学阴极还原活化后其表面出现了很多三角形或者截角三角形的凹坑,如图7b 所示。进一步的EDX 点分析(TEM 模式,spot size 6, 空间分辨率约1~2 nm)显示(图7(c-d)),凹坑处的S∶Nb 的比值约为1.1,而完整的地方的比值为2.0,表明电化学处理使其表面的S 成片地消失了。这与此前在电化学活化的MoS2中的单个S 空位结构显著不同。
为了确认其活化带来的结构差异,采用原位电化学拉曼光谱仪研究了3 种TMDs 在析氢反应不同电势下的结构变化。所测试的电压区间为−0.6~0 V(vs.RHE)。对于MoS2,发现其在不同电压下,表征其结构的3 个主峰特征峰的、A1g、C 峰位和峰强几乎无变化,(图8b)其余8 个小峰也都没有变化(图8a)[31];WS2的拉曼峰的位置和强度都没有变化(图8d)[32]。这表明在析氢电位下MoS2和WS2是相对比较稳定的,所形成的原子级的硫空位并未影响整体结构的拉曼散射,振动模式没有变化即主体结构没有明显变化。对于NbS2(图8c),原始NbS2(0 V 电位下的拉曼光谱)的四个主要峰位E1(280 cm−1)、E2(326 cm−1)、A1(386 cm−1)、A2(456 cm−1)与3R 相NbS2对应,尖锐的A1峰也表明了结晶性较好[33]。随电势降低,其E2峰稍向高位移处偏移,且A1的强度在减弱,(图8c)尤其是在224 cm−1处出现了新的振动峰[33,34],说明NbS2相对更不稳定,大量相邻的S2-与H2结合生成H2S。这与前面图7a 中出现大量的大面积S 空位区一致,也可以解释为何NbS2经过电化学活化后,其产氢性能的提高幅度明显小于MoS2活化所带来的性能提升。
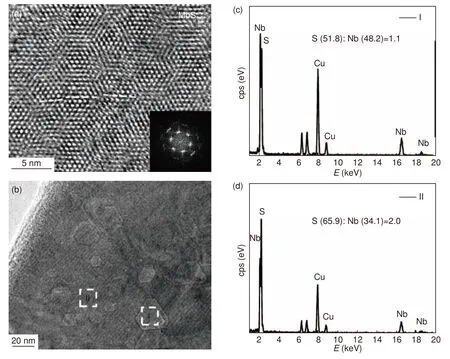
图7 (a)活化后的MoS2 的HRTEM 照片及该区域的FFT,(b)活化后的NbS2 的TEM 照片以及(b)中两个区域(c)I 和(d)II 的EDX 谱图Fig.7 (a) HRTEM image of activated MoS2 with FFT, (b) TEM image of activated NbS2, EDX patterns of two regions (c) I and (d) II from Fig.7(a)
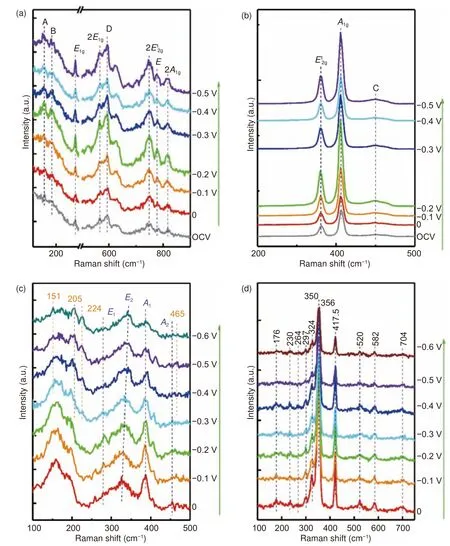
图8 三种TMDs 在0~ –0.6 V(vs.RHE)电位下的原位电化学拉曼光谱图:(a-b)MoS2、(c)NbS2、(d)WS2Fig.8 In-situ electrochemical Raman spectra of (a, b) MoS2, (c) NbS2 and (d) WS2 under steady state chronoamperometry by holding at each potential (from 0 to -0.6 V/RHE) for 2 min
4 结论
以导电性优异的炭纸作为衬底,利用化学气相沉积方法在其表面生长了3 种TMDs 纳米片,不使用黏结剂而直接作为产氢电催化反应的一体化电极。系统探索了炭纸表面生长3 种直立型二维TMDs (MoS2,NbS2以及WS2)的反应温度和气体流量对成分和结构形貌的影响。实验发现反应温度对所合成的二维过渡金属硫化物的成分有显著影响,但对不同过渡金属硫化物影响的趋势不同。气体流量则对样品的产率以及形貌有较大影响,且气体流量越大,样品的产率普遍越高。所制备的3 种TMDs 中,WS2表现出一种新颖的纳米片一维组装结构,且纳米片的平面尺寸在3 种TMDs 中最小,暴露出的边缘位最多,因此表现出最佳的产氢性能。进一步实验证实阴极电化学活化可以有效提高TMDs 的HER 活性。结合TEM 和原位电化学拉曼光谱图发现,与之前报道的活化后的MoS2中出现大量单个S 空位缺陷不同,NbS2在阴极活化后出现了大片的三角形或者截角三角形S 空位区,从而其性能的提升幅度相对较小。
致谢
感谢国家自然科学基金(No.52173222);广东珠江人才计划地方创新研究团队项目(No.2017BT01N111);广东省自然科学基金杰出青年基金(No.2016A030306035)。
猜你喜欢
杂志排行

新型炭材料的其它文章
- Ni(OH)2/石墨相氮化碳/石墨烯三元复合材料的制备及电化学性能
- Coal-based graphene as a promoter of TiO2 catalytic activity for the photocatalytic degradation of organic dyes
- Preparation and lithium storage of anthracite-based graphite anode materials
- A flexible hard carbon microsphere/MXene film as a high-performance anode for sodium-ion storage
- 咖啡渣成型制备生物质炭及其CH4/N2分离性能
- Oxygen-incorporated carbon nitride porous nanosheets for highly efficient photoelectrocatalytic CO2 reduction to formate