钢中NbC 夹杂粒子表面性质的第一性原理
2022-12-02李云阔武英举梅国宏张庆军2
陈 伟, 李云阔, 李 耀, 武英举, 梅国宏, 陈 颖, 张庆军2,
(1. 华北理工大学 冶金与能源学院, 河北 唐山 063210;2. 河北省高品质钢连铸工程技术研究中心, 河北 唐山 063000;3. 华北理工大学 综合测试分析中心, 河北 唐山 063210;4. 燕山大学 亚稳材料制备技术与科学国家重点实验室, 河北 秦皇岛 066004)
NbC 作为一种重要的岩盐型过渡金属碳化物,具有高熔点、超高硬度和优异的化学稳定性[1].以大线能量焊接船板钢DH36 为例,在低温铁素体相变过程中NbC 粒子可大量析出[2],其中附着在铁素体晶面的粒子具有钉扎作用,可抑制铁素体晶粒长大,促使大量针状铁素体生成,从而使晶粒尺寸进一步细化[3-4].对NbC 粒子表面性质的研究,有助于揭示针状铁素体的形成条件及生长方式.
近年来,密度泛函理论(DFT)开始被用于纳观材料表面性质的计算.张怀征、Xiong 等[5-6]对NbC(001)面的表面能、表面褶皱性质及电子态进行了研究,发现表面能可从平板的总能量中减去体相能量求得.Liu 等[7]通过模拟计算,得到NbC低指数(001)面是最稳定表面.Yang 等[8]计算了NbC(111)面的表面能,利用表面能的收敛性确定了最小原子层厚度.章永凡等[9]对NbC(111)面的电子结构进行了理论研究,对弛豫前后的原子结构进行了相应探讨.有关NbC 表面性质的研究刚刚起步,NbC 所有低指数面的表面特性还需要进行系统的分析.
本文中利用基于密度泛函理论的第一性原理平面波(动态生成)超软赝势法,对NbC 四种低指数面即(001)面、(110)面、Nb 终止(111)面及C终止(111)面的表面性质进行系统研究,从而对基态结构进行更加精确的描述.考虑到碳化学势对表面能的影响,在计算极性(111)面表面能时,引入了Nb 及C 单质化学势等影响因素,使计算结果更加合理.文中较全面地分析了NbC 的表面结构、表面能及电子结构等表面性质对表面稳定性的影响.
1 计算方法和模型
本文中利用CASTEP 软件进行第一性原理的计算与分析.其中电子交换关联能采用广义梯度近似(GGA)中的PBE 函数进行修正[10];通过动态生成的超软赝势(OTFG ultrasoft)描述价电子与离子实的相互作用,Nb 及C 原子的价电子分别为4s24p64d45s1与2s22p2; NbC 平面波截断能选取600 eV;在自洽场运算中,采用Pulay 密度混合法[11],自洽场(SCF) 收敛精度设置为每个原子2×10-6eV;由于Nb 是顺磁性金属,NbC 低指数面结构弛豫计算未考虑自旋极化;采用BFGS(Broyden-Fletcher-Goldfarb-Shannon) 算 法[12],对晶胞进行几何优化;K 点的选择采用Monkhorst-Pack 网格划分方法[13],对布里渊区进行积分,表面采用[9×9×1]的K 点网格进行划分;结果分析时,利用Mulliken 电荷布局分析方法[14-15],对体系原子的电荷量进行计算.在表面结构弛豫优化过程中,对所有原子均进行自由弛豫,以考察结构中各层原子的表面效应.
NbC 晶体结构为面心立方结构,如图1 所示,其低指数面原子结构如图2 所示,分别为(001)面、(110)面、Nb 终止(111)面及C 终止(111)面.表面结构选用(1×1)超级晶胞进行计算;真空层厚度设置为1.2 nm,可以有效避免相邻单元结构间的原子相互影响.

图1 NbC 晶体结构示意图Fig.1 Schematic diagram of NbC crystal structure

图2 NbC 低指数面原子结构Fig.2 Atomic structure of NbC low-index surface
2 结果与讨论
2.1 表面结构弛豫
为了使结构内部既具有体相特性,又节省计算成本,需要对原子层数进行收敛性测试,以确定最小原子层数.四种低指数面分别采用含3 层、5层、7 层、9 层、11 层、13 层原子的对称结构进行讨论,避免极性面带来的偶极矩效应.
由于晶体具有周期性和对称性,NbC 各表面原子在水平方向上很容易达到力的平衡,发生的相对位移较小,可以忽略不计;在垂直方向上,晶体的周期性会因表面的形成而中断,导致表面原子受力失衡,使Nb 原子、C 原子在垂直于表面的方向上发生偏移.NbC(001)及(110)面经过结构弛豫后,由于原子受力不同,表面均出现了褶皱,使得每层的C 原子与Nb 原子不在一个平面上.鉴于此,原子层平面纵坐标采用各原子层的C 与Nb原子纵坐标的平均值来表示.NbC(001)表面层的C 原子向真空层一侧偏移,而Nb 原子向体相一侧偏移,弛豫程度随着原子层数的增加而变小,逐渐趋于稳定.NbC(110)面最外层的Nb 及C 原子均向体相一侧偏移,但偏移程度不同,这主要是由于离子半径不同,最外层Nb 原子的弛豫程度要大于C 原子的弛豫程度.Nb 终止(111)面及C 终止(111)面经过弛豫后,最外两层的原子层间距亦出现了缩小的现象,随着原子层数的增加,层间距逐渐收敛并趋于稳定,最外层的Nb 及C 原子均向体相一侧移动.
将NbC 不同表面结构层间距随原子层数(n)的变化列于表1~3 中(表中Δij代表弛豫前后层间距的变化相对于体相层间距的百分比).从表中可以看出,随着原子层数的增多,原子层间距也在逐渐趋于收敛,当(001)面、(110)面、Nb 终止(111)面及C 终止(111)面的层数分别取值5,9, 13, 11 时, 结构中出现体相特征. 通过NbC各低指数面的弛豫情况可以看出, NbC(001)面的结构弛豫效果最不明显,而C 终止(111)面的结构弛豫效果最为显著.

表1 NbC(001)面结构弛豫结果Table 1 NbC(001) surface configuration relaxation results %

表2 NbC(110)面结构弛豫结果Table 2 NbC(110) surface configuration relaxation results %

表3 Nb 及C 终止(111)面结构弛豫结果Table 3 Nb and C termination (111) surface configuration relaxation results %
2.2 表面能的计算
为了比较NbC 各低指数面的相对稳定性,对其表面能进行了计算.其中NbC(001)面与(110)面均为非极性面,因此表面能σ 计算采用如下公式[16]:
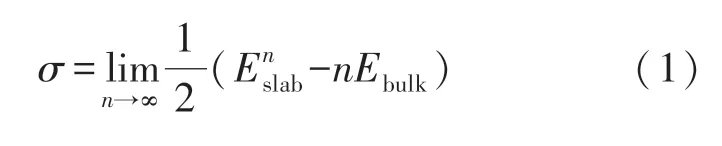
式中:n 代表原子层数;“1/2”代表平板具有两个表面; Enslab代表弛豫后具有n 层原子平板的体系总能量; Ebulk代表体相系统的能量.
由于Nb 终止及C 终止(111)极性面采用了对称结构,表面能会随着化学势的改变而变化.因此,在计算Nb 及C 终止(111)面的表面能时,应考虑Nb 及C 原子化学势对表面能的影响.二者的表面能公式可表示为[17-18]

式中: A 代表结构表面的面积; Eslab代表弛豫后Nb 终止及C 终止(111)面结构的总能; NNb和NC分别代表Nb 终止及C 终止(111)面超级晶胞Nb原子和C 原子个数; μbNublk和μbCulk分别代表Nb 与C的体相化学势; p,V,T,S 分别代表体系的压强、体积、温度及熵.在温度为0 K 及恒定压力的条件下,pV 和TS 值可忽略不计.表面结构经过充分优化弛豫后,内部结构处于平衡状态.NbC 的体相化学势μbulkNbC可以表示为

式(2)可以改写为含有μslab
C 项的函数形式:

此外,基于上述公式转换, NbC 的体相化学势与NbC 在0 K 时的形成焓ΔH0f以及Nb,C 在单质相中的化学势μbulkNb, μbulkC有关,可表示为

式(3)及式(5)联合可得:

式中, μbNublk和μbCulk可分别通过对体心立方结构的Nb 单质、金刚石结构的C 单质计算得到.另外,表面结构中Nb 及C 的化学势必须分别小于其在单质体相中的化学势[19].否则,NbC 表面结构将不稳定且易发生分解,转变为能量更小且更稳定的单质Nb 和C.因此,从热力学角度出发,Nb 的化学势取值范围应满足下面的关系式:

经计算,ΔH0f为-1.27 eV,与文献值[20-21]吻合良好.
图3 为Nb 终止(111)面和C 终止(111)面表面能随C 化学势(μsClab-μbCulk)的变化关系图.为了比较各低指数面的相对稳定性,将NbC(001)及(110)面的表面能与Nb 终止及C 终止(111)面的表面能同时显示在图中.由图可知:NbC(001)面与(110)面的表面能不会随C 化学势的变化而改变(图中以水平直线的形式显示);而Nb 终止(111)面及C 终止(111)面的表面能则与C 化学势呈线性相关,Nb 终止(111)面的表面能会随C化学势的增加而线性增大,C 终止(111)面的表面能会随C 化学势的增加而线性减小.比较整个C 化学势取值范围内低指数面表面能的大小后发现,C 终止(111)面表面能最大,(110)面与Nb 终止(111)面表面能次之,(001)面的表面能最小,仅为1.31 J/m2.因此,NbC(001)面的热力学稳定性最好,可以稳定存在.

图3 NbC 低指数面表面能与C 化学势之间的关系Fig.3 The relationship between the surface energy of the low-index surface of NbC and the chemical potential of C
2.3 表面电子结构及成键特性
本节中利用Mulliken 电荷布局分析方法, 分别对NbC 各低指数面弛豫前后的有效电荷转移量进行了计算, 并将弛豫前后的电荷转移情况和相邻原子层且不同原子之间化学键键长列于表4~6中. 从表中可以看出, 电荷发生转移、 原子之间电荷重新分布使得原子间出现静电斥力, 引起表面垂直方向上相邻Nb 及C 原子之间键长变化. 比较弛豫前后的化学键键长后发现: NbC(001)面第一层的C 原子与第二层的Nb 原子的化学键键长增大, 第一层的Nb 原子与第二层的C 原子的化学键键长减小, 与结构中Nb 及C 原子分别向体相一侧或真空层一侧偏移对应, 进一步说明NbC 表面存在褶皱; NbC(110)面第一层C(Nb)原子与第二层Nb(C)原子的化学键键长均减小, 第一层的Nb 原子相较于C 原子而言向体相一侧偏移程度更大; NbC(111)面的化学键键长的变化随原子层数的增加而逐渐趋于稳定.
对比不同的表面结构后发现, Nb 原子与C原子之间有效电荷的转移量各不相同. NbC 各低指数面中的C 终止(111) 面弛豫幅度最大, 有效电荷转移量最多, 为0.12 e. NbC(001)面弛豫幅度最小, 有效电荷转移量最少, 为0.01 e. 比较Nb 终止(111) 面和C 终止(111) 面的化学键键长变化情况, 发现C 终止(111) 表面化学键键长变化更显著. 由此可知, NbC 的C 终止(111) 表面原子间共价键更强, 进而导致了层间距的显著变小.

表4 NbC(001)面原子所带电荷及原子间键长Table 4 The charges on the atoms of NbC(001) surface and the bond length between atoms
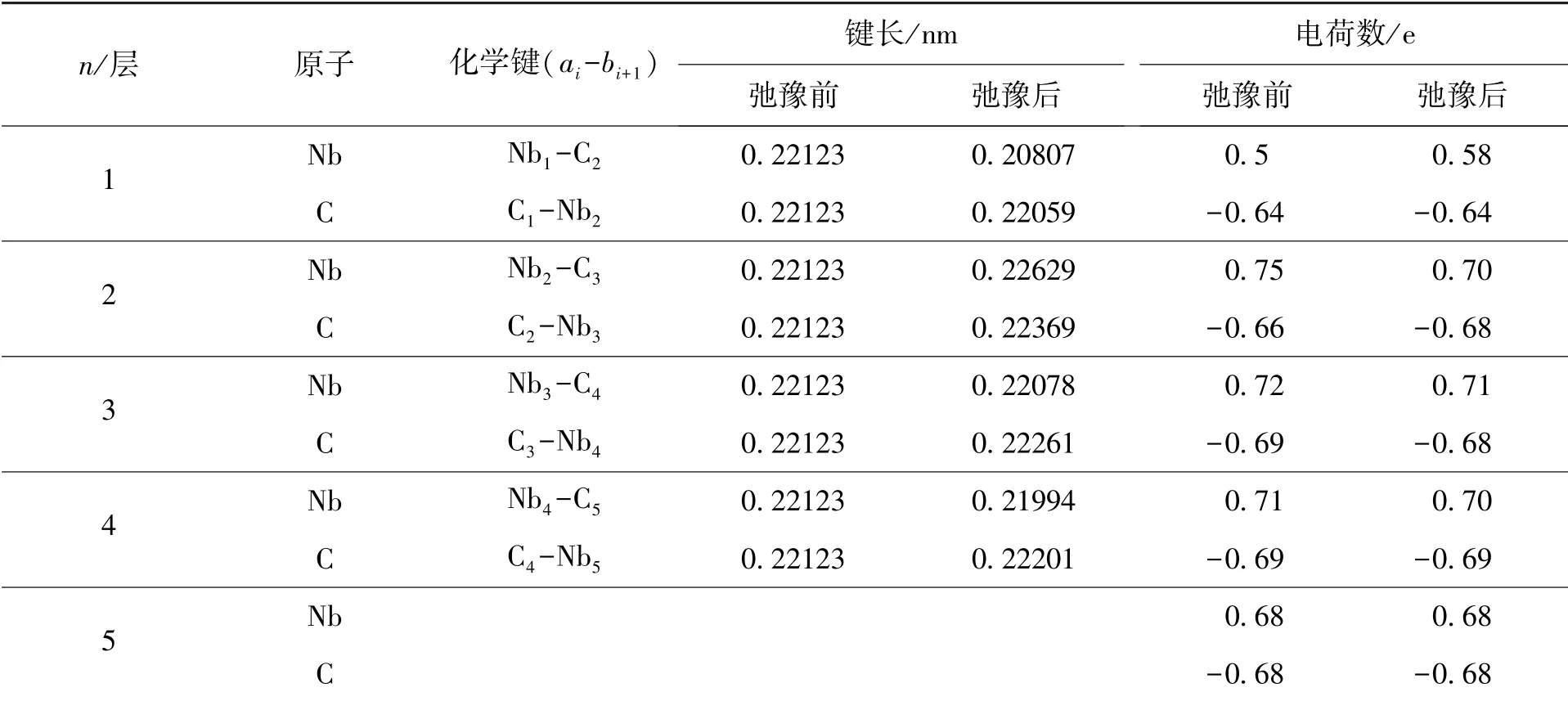
表5 NbC(110)面原子所带电荷及原子间键长Table 5 The charges on the atoms of NbC(110) surface and the bond length between atoms

表6 Nb 及C 终止(111)面原子所带电荷及原子间键长Table 6 The charges of atoms on the (111) surface terminated by Nb and C and the bond length between atoms
2.4 NbC 各低指数面的态密度分析
为了进一步分析NbC 各低指数表面的化学键,分别对结构弛豫后的NbC 体相以及各低指数面总态密度(DOS)特性进行计算.Nb 与C 的价电子分别为4s24p64d45s1和2s22p2,后续偏态密度(PDOS)的计算主要针对能级较高的Nb-4d 轨道以及C-2s,C-2p 轨道进行.图4 为NbC 的体相以及各低指数面的态密度对照图,图中费米能级(0 eV)处用虚线标注.
从图中可以看出,在-13 ~-8 eV 能量区间的态密度主要由C-2s 轨道组成,并且掺入了部分Nb-4d 轨道的贡献.在-7.5 ~0 eV 能量区间的态密度大部分区域为Nb-4d 态和C-2p 态之间杂化作用形成的成键态,仅有少部分Nb-4d 态和C-2p态在靠近费米能级一侧区域杂化形成反键态;大部分反键态位于费米能级上方.
观察各低指数面偏态密度图[图4(b) ~(e)]并比较费米能级下方的反键态区域后发现,C 终止(111)面的反键态区域最小,降低了该能级处的电子填充到反键态上的概率,从而对化学键成键能力的削弱作用也最小[5].同时,在C 终止(111)面的成键态处,Nb-4d 态与C-2p 态有明显的重合,存在剧烈的轨道杂化作用,产生了强烈的共价键.这都说明了结构弛豫优化后,C 终止(111)面具有最大的弛豫幅度.

图4 NbC 体相及各低指数面的态密度Fig.4 Density of state of NbC bulk phase and low-index surfaces
3 结 论
(1)对NbC 低指数面结构弛豫优化后,得到各低指数面[(001)面、(110)面、Nb 终止(111)面、C 终止(111)面]具有体相特征的最小原子层数分别为5 层、9 层、13 层和11 层,弛豫效应主要发生在表面前三层,其中NbC(001)面的结构弛豫效果最不明显,C 终止(111)面的结构弛豫效果最为显著.
(2)计算得到NbC 各低指数面表面能,其稳定性排序为C 终止(111)面<(110)面<Nb 终止(111)面<(001)面;(001)面的表面能最小,为1.31 J/m2,但结构最稳定.
(3)对低指数面的电子结构以及键合特性进行研究后发现,(001)面的有效电荷转移量最小,仅为0.01 e,C 终止(111)面的有效电荷转移量最大,为0.12 e.结构弛豫优化后,Nb 原子与同层或相邻层C 原子之间形成了强烈的极性共价键,相邻Nb 原子之间也形成了较强的金属键,与原子层的弛豫效应对应一致.从态密度图中可知,C 终止(111)面中Nb 原子与C 原子之间的键合能力最强,这与原子层的弛豫效果相吻合.
(4)对NbC 各低指数面稳定性、电子结构以及键合方式的理论分析,有助于揭示NbC 夹杂粒子的表面特性,对探讨NbC 与钢基体间的界面结合方式以及位向关系有一定的指导意义.
