构建人源化CYP4V2点突变小鼠的CRISPR/Cas9系统△
2022-11-25赵莹科方源钱丽玲王伟吴继红
赵莹科 方源 钱丽玲 王伟 吴继红
(1.复旦大学附属眼耳鼻喉科医院眼科 上海 200031; 2.上海市视觉损伤与重建重点实验室 上海 200031;3.国家卫生健康委员会近视眼重点实验室 上海 200031; 4.中国科学院脑科学与智能技术卓越创新中心 上海 200031)
结晶样视网膜变性(Bietti crystalline corneoretinal dystrophy,BCD)是一类常染色体隐性视网膜营养不良症,临床表现为视网膜上黄白色脂质结晶沉积[1],伴有视网膜色素上皮萎缩、脉络膜硬化,进而出现视力下降、夜盲、视野丧失和色觉受损,甚至发展为失明。尽管在欧美人群中也有对于BCD的报道,但是BCD在亚洲人群中的发病率更高[2]。Li等[3]通过对25例患者进行基因关联分析,首次确定染色体4q35是BCD患者遗传缺陷的高发区域,并在23例患者中发现了CYP4V2基因的突变。
CYP4V2基因共包含11个外显子,编码的蛋白属于细胞色素P450家族的成员。属于P450家族4的一员,表明CYP4V2是脂肪酸ω-羟化酶。CYP4V2被认为能够催化中等链长的脂肪酸,肉豆蔻酸、月桂酸、棕榈酸和多不饱和脂肪酸(PUFAs),后者包括花生四烯酸(A.A.)、二十二碳六烯酸(DHA)、二十碳五烯酸(EPA)等[4]。目前主要认为CYP4V2可以影响脂肪酸代谢,但是CYP4V2的特异性底物尚不清楚,突变如何干扰脂肪酸代谢从而引起视网膜变性的具体作用机制也未明确。
基因修饰动物模型是在活体动物上开展基因功能研究、寻找合适药物作用靶点的重要工具[5]。尽管既往已经有许多学者[6]报道了CYP4V3基因敲除鼠的相应表型及可能存在的机制,但是仍无法完全反映人类BCD的病变特征。因此,基于人的突变位点构建人源化动物模型,对于BCD的发病机制以及基因治疗药物的开发,意义重大[7]。

表 1 用于构建供体质粒的PCR引物序列
1 材料与方法
1.1 人CYP4V2致病位点的筛查 首先利用gnomAD在线数据库检索CYP4V2功能失活突变;同时利用ClinVar数据库检索CYP4V2致病突变情况。综合分析,确定人CYP4V2明确的高频强致病变异位点。
1.2 线性供体的构建与鉴定 确定突变位点后,从人的胚胎干细胞基因组上选取突变区域共408 bp的序列(含1091-2 A>G),以及该突变前910 bp,还有供体左右同源臂(HAL:801 bp; HAR:820bp),应用Tild-Crispr方法构建供体质粒(图1),具体构建方法和同源臂设计见文献[8]。供体质粒的聚合酶链反应(polymerase chain reaction,PCR)引物序列如表1所示。
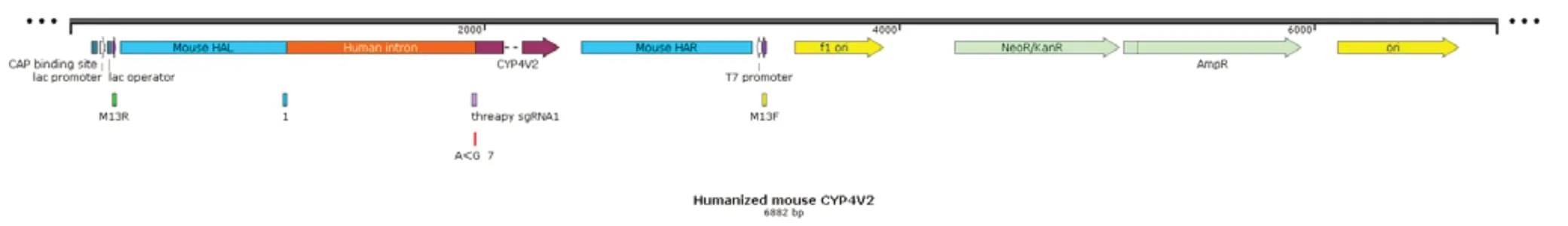
图 1 CYP4V3人源化小鼠供体质粒图谱
1.3 Cas9 mRNA与sgRNA的构建 将T7启动子以及Cas9的编码区通过在px260载体进行连接,PCR产物经过纯化,使用mMESSAGE mMACHINE T7(Life Technologies)体外转录试剂盒进行体外转录,得到Cas9 mRNA。此外,依据tild-CRISPR已发表的方法构建sgRNA模板,通过与Cas9 mRNA类似的体外转录方法,得到sgRNA。Cas9 mRNA和sgRNA产物均使用MEGA clear (Life Technologies)试剂盒进行纯化。
1.4 小鼠受精卵显微注射 超排雌性C57BL/6小鼠(7~8周龄)通过先注射5 IU孕马血清促性腺激素(PMSG)、48 h后注射5 IU人绒毛膜促性腺激素(human chorionic gonadotrophin,hCG)与雄性小鼠交配,并在注射hCG后20 h从输卵管收集受精胚胎。使用具有恒定流量设置的FemtoJet显微注射器将Cas9 mRNA、sgRNA和线性供体质粒在含有5 μg/mL松弛剂的液滴中混合并注射到小鼠受精卵中。
1.5 小鼠基因型鉴定 小鼠出生1周后,使用TIANamp Genomic DNA Kit (TIANGEN,DP304-03)从脚趾或尾巴样本中提取小鼠基因组DNA。使用设计用于扩增正确靶向连接的引物进行PCR扩增(表2、3)。ExTaq在95 °C下激活3 min,然后在 95 °C 30 s、60 °C 30 s和 72 °C 1 min进行 38 个循环,最后在 72 °C下延伸 10 min。PCR产物经凝胶纯化和测序。

表 2 用于鉴定疾病模型小鼠是否携带供体片段的PCR引物序列

表 3 用于鉴定携带1091-2 A>G突变的CYP4V3人源化小鼠CYP4V3 mRNA splice mutant的PCR引物序列
2 结果
2.1 人CYP4V2致病位点的筛查结果 结合gnomAD与ClinVar上关于质变变异的突变数据,gnomAD共收录860种突变,其中明确致病的突变有24种;ClinVar数据库中共检索到560个突变信息,其中致病突变144个,30个致病位点与BCD相关。将gnomAD与ClinVar的致病信息取交集,得到12个变异位点,认为这12个是较为明确的致病位点(表4)。根据课题组既往对于基于我国眼遗传病患者基因突变情况的临床研究,最终选定CYP4V2c.1091-2 A>G作为后续人源化动物模型的致病突变位点。在Ensmbl数据库中导出小鼠CYP4V3基因序列,与人CYP4V2基因进行比对,发现人CYP4V2c.1091-2 A>G基因与小鼠相应区域序列一致性较高(图2)。
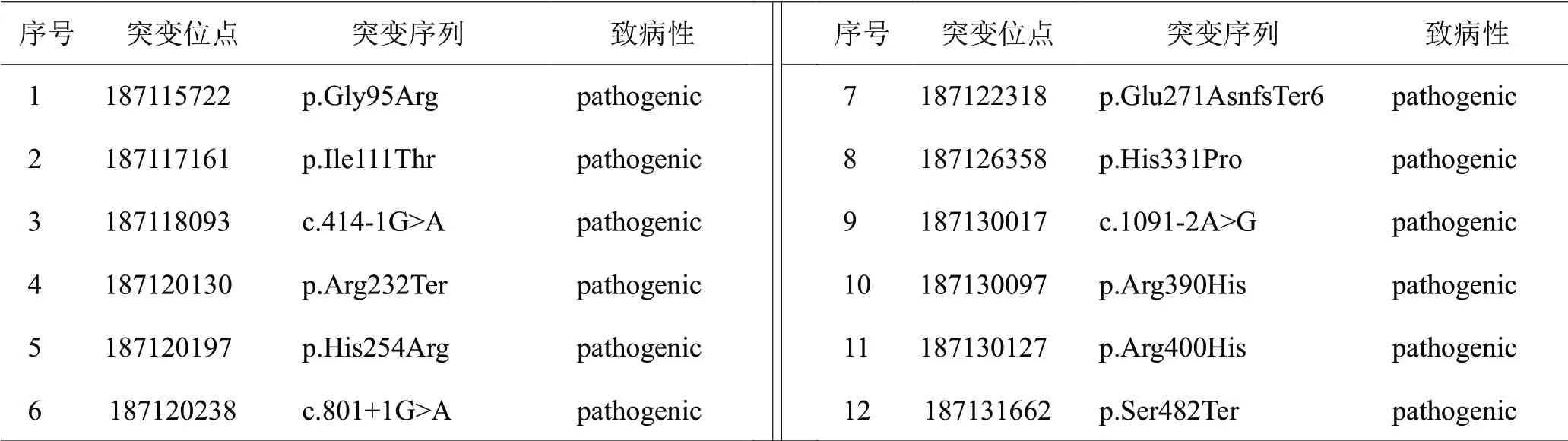
表 4 gnomAD与ClinVar中收录的CYP4V2致病突变情况
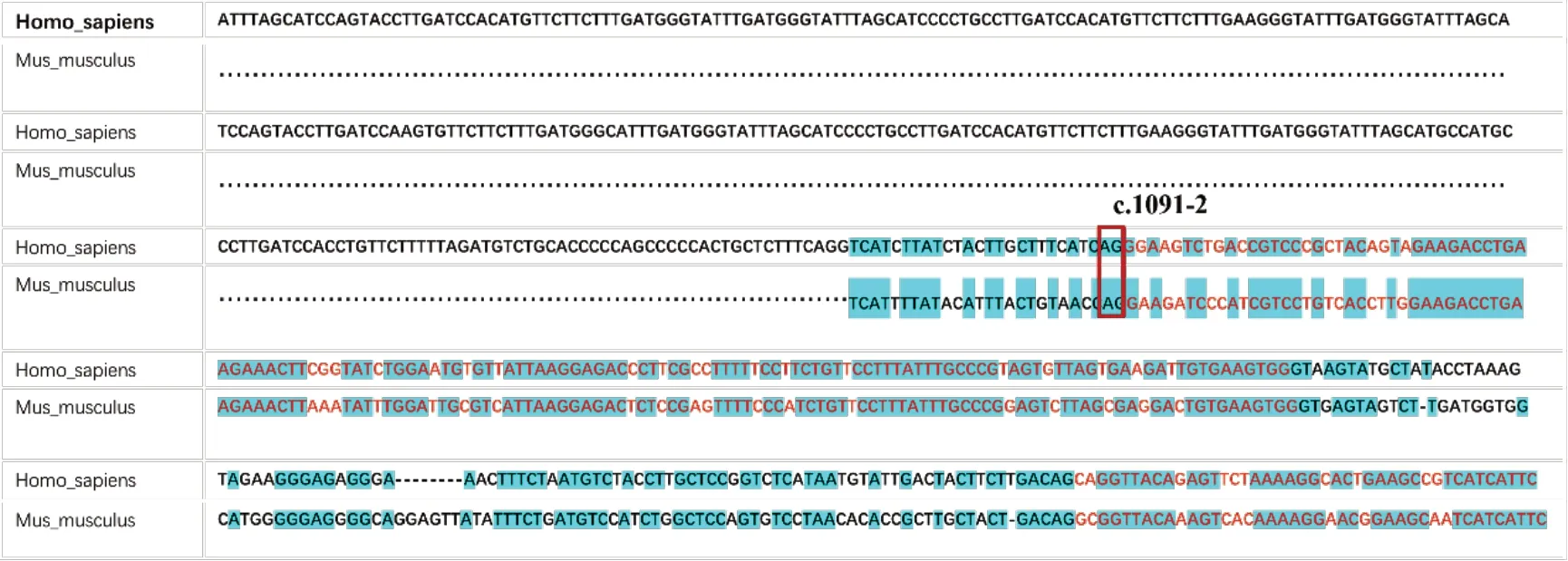
图 2 人CYP4V2与鼠CYP4V3基因序列的比较 其中红色碱基代表外显子区域,蓝色标志为人、鼠一致的基因序列。红框所示c.1091-2,为本研究确定的突变区域,可见人、鼠在该区域的序列一致。
2.2CYP4V3人源化小鼠在DNA水平构建成功 如图3所示,通过二代测序结果可以确定,构建完成的基因编辑鼠在CYP4V31091-2的位置发生了A>G的突变。携带1091-2 A>G突变的CYP4V3人源化小鼠参考序列包括人工插入替换的携带1091-2 A>G突变人源内含子部分以及人源CYP4V2中原本具有的第11个外显子部分。经测序,阳性小鼠的序列完全符合阳性小鼠参考序列的碱基排列,1091-2位置的A已经突变为G,并有干净稳定的峰图;而野生型作为对照,不含有人工插入替换的人源内含子部分,但含有野生型小鼠CYP4V3基因第11个外显子部分。
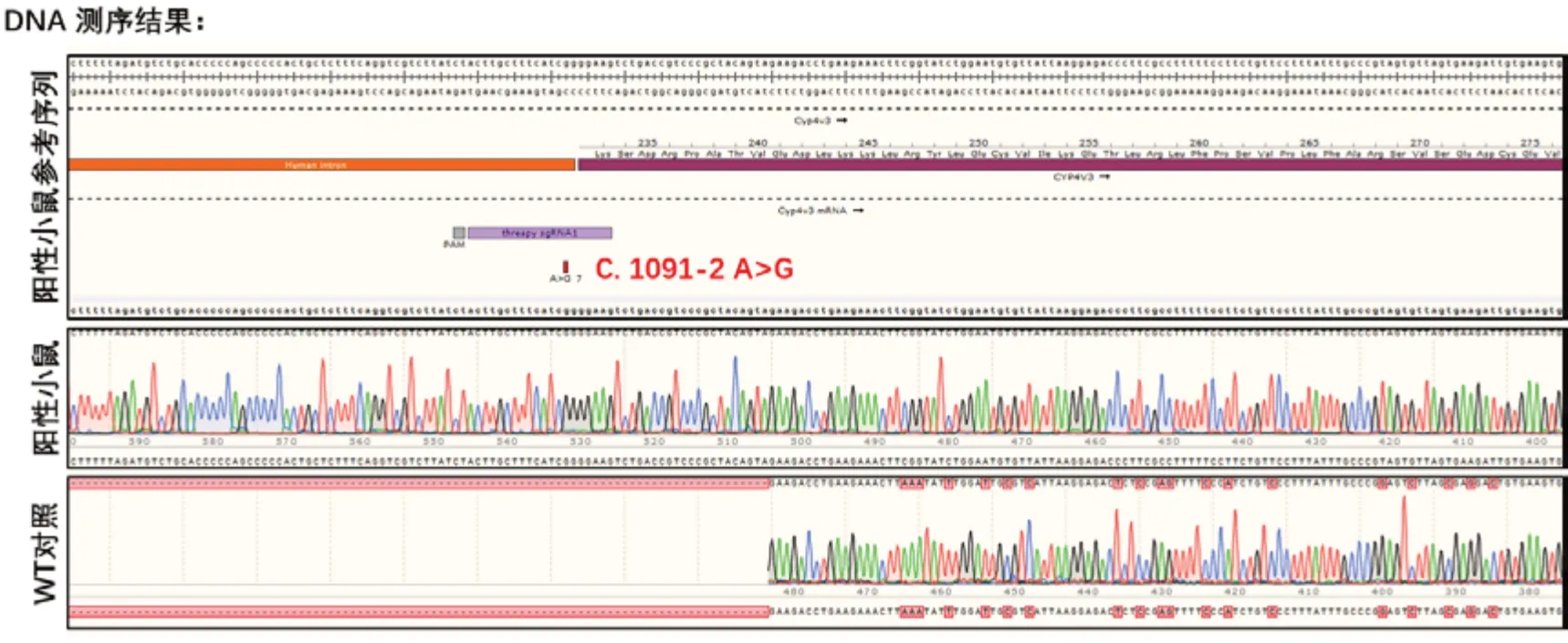
图 3 携带1091-2 A>G突变的CYP4V3人源化小鼠在DNA水平上构建成功
为了保证得到的小鼠模型能够模拟人体内的情况,为今后的临床筛查做准备,我们将小鼠CYP4V3基因的第9和第10个外显子,包括其中的内含子,完全替换成了人源。由于1091-2 A>G突变位点正好在人第9个外显子前的内含子的碱基最后一位,我们将第9个外显子前的内含子后910 bp也替换成了人源内含子。因此,一共需要将910 bp携带点突变的内含子及其后的第9、10两个外显子替换成人源。确定好替换方案后,在需要替换的基因前后分别设计了4个sgRNA识别位点,体外验证这8个sgRNA的工作效率,希望通过基因编辑的方式将野生型小鼠里的这段整段敲除,并替换成携带1091-2 A>G突变的CYP4V3人源化片段。
经过对插入片段的完整测序,我们确认插入段碱基与阳性小鼠参考序列对比一致,且测序峰图干净单一,排除因测序过程带来的实验误差。此外,我们用野生型小鼠对比测序,发现野生型小鼠没有插入片段,且CYP4V3基因的第9个外显子与阳性小鼠参考序列断断续续有部分碱基重合,证明小鼠和人的CYP4V3基因第9个外显子有一定的同源性。至此,我们在DNA水平上证明了携带1091-2 A>G突变的CYP4V3人源化小鼠构建成功。
2.3 无法判断携带1091-2 A>G突变的CYP4V3人源化小鼠CYP4V3mRNA 是否产生剪切突变 根据临床上的报道[9],CYP4V2基因上携带有1091-2 A>G突变患者的mRNA在加工过程中会产生剪切突变,从而跳过整个第9号外显子,最终导致疾病的发生。如图4所示,我们取构建完成的基因编辑鼠视网膜处的总RNA进行反转录,并对得到的cDNA进行测序后发现,构建完成的小鼠会在小鼠第8号外显子和人的第9号外显子中间产生无规则序列,并导致后续一系列碱基异常表达,既无法判断其是否跳过了第9号外显子导致疾病的产生,也无法判断其是否能够让第8、9号外显子正常表达。

图 4 携带1091-2 A>G突变的CYP4V3人源化小鼠cDNA测序结果
3 讨论
本研究中,我们通过应用Tild-CRISPR技术,成功将携带1091-2 A>G突变的人CYP4V2基因片段408 bp导入了小鼠CYP4V3基因,DNA测序结果提示,本次实验成功地引入了1091-2 A>G突变,对于后续CYP4V2基因的研究具有重要的借鉴意义。
尽管CYP4V2基因全身均有表达,但是目前数据库中记录CYP4V2基因突变仅引起眼部病变,由于CYP4V2蛋白活性异常导致的脂肪酸代谢紊乱并不能完全解释这一现象,因此探索其发病机制、寻找有针对性的治疗手段,阻断这一常见致盲性眼病进程,具有重要的临床价值。通过诱导CYP4V2突变患者的干细胞并分化形成视网膜色素上皮层(retinal pigment epithelium,RPE),Hata等[10]报道了CYP4V2突变的RPE细胞中葡糖苷酰鞘氨醇(GlcCer)的累积以及胆固醇酯的减少。这与此前研究人员在CYP4V2突变患者成纤维细胞上,应用放射性同位素标记观察到脂肪酸向n-3多不饱和脂肪酸转化减少[11],共同说明了CYP4V2突变可引起了体内脂质代谢的多个环节异常,破坏了细胞内的脂质稳态从而导致疾病的发生[12]。但是CYP4V2突变产生的具体分子生物学效应,目前仍不清楚。因此我们应用CRISPR/Cas9基因编辑技术,开展了对携带人CYP4V2热点突变的模式动物的探索。
CRISPR/Cas9介导的基因编辑技术的发展,使得组织、细胞基因水平的原位纠正变为现实,也推动了基因治疗领域的极大发展。Cas9切割后产生的双链断裂,在没有模板存在的情况下,会利用非同源末端连接的方式进行修复;而在有修复模板的时候,DNA断端可以通过高保真的同源重组方式,将模板DNA序列精确插入到目标DNA序列中。非同源末端连接的发生率非常高,而同源重组的发生率较低。tild-CRISPR是由中国科学院脑科学与智能技术卓越创新中心杨辉教授团队报道的一种新型基因打靶策略,其优势在于更高的DNA敲入效率[8]。同时对于插入片段的大小从800 bp到6 000 bp都能够精确整合到不同的位点。与以往的基因编辑方式不同,tild-CRISPR采用的是线性的DNA供体,可以显著提高基因编辑的效率。在本研究中,我们应用tild-CRISPR成功替换了小鼠基因组上的408 bp。DNA水平,小鼠CYP4V3基因序列中成功实现了携带人源1091-2 A>G点突变序列的同源定点替换;RNA水平,利用反转录PCR分析cDNA序列,发现突变小鼠cDNA在1091-2 A>G点突变序列附近发生了错误的可变剪切,这与临床上携带1091-2 A>G点突变的患者表型一致。然后在后续的表型鉴定中,我们并没有发现与预期一致的蛋白水平表达变化(数据未展示),提示错误的可变剪切可能并未改变CYP4V3总体的蛋白水平。考虑到该病的发病周期个体差别较大,未来我们将对模型进行更长周期的观察以及表型的鉴定。
综上所述,我们利用CRISPR/Cas技术,成功制备了携带患者特异性遗传突变的小鼠模型,为以后深入探索CYP4V21091-2 A>G点突变的致病机制以及开发相应的基因治疗药物奠定了基础。
