遗传性视神经病变概述
2022-11-25田国红
田国红
[1.复旦大学附属眼耳鼻喉科医院眼科 上海 200031; 2.国家卫健委及中国医学科学院近视眼重点实验室(复旦大学) 上海 200031; 3.上海市视觉损害与重建重点实验室(复旦大学) 上海 200031]
1 Leber遗传性视神经病变
1871年,Leber首次描述了该病并冠名[1],但直到20世纪80年代才明确该病为母系遗传的线粒体基因突变导致。线粒体基因为独立于核基因之外的双链DNA环,位于真核生物细胞质中,负责编码真核生物氧化呼吸链不可缺少的还原性辅酶Ⅰ脱氢酶复合体(NADH),该复合体的3个亚单位ND4、 ND6和ND1分别由线粒体DNA(mtDNA)11778位点、14484位点和3460位点编码。如果上述3个位点出现点突变则复合体功能受损而无法产生三磷酸腺苷(ATP)为细胞供氧[2]。
流行病学数据显示,在新英格兰人群中,Leber遗传性视神经病变(Leber hereditary optic neuropathy,LHON)的发病率为3.22/100 000[3]。目前缺乏国内流行病学数据。在具有基因突变的家系中,男性患者出现视力下降的比率可高达60%,而女性约30%发病。有症状的女性更容易生育有症状的下一代[4]。部分携带mtDNA突变的患者可以终身无症状,可能与个体突变的杂合度、基因表型及缺陷基因的拷贝数、组织的需氧量、外界诱发因素相关。由于突变的mtDNA存在于胞质中,生殖过程中精子中所含线粒体进入受精卵的量很少,此为LHON母系遗传的分子生物学基础。
1.1 临床特征 患者多为15~35岁的青少年,男性多见。临床中老年患者亦可见。以急性或亚急性无痛性视力下降为常见就诊主诉。双眼可同时或先后受累。两眼间隔发病时间常为数周至数月,且97%的患者对侧眼在1年内出现视力下降。急性期患眼视力呈持续性下降的趋势,直至3个月左右趋于稳定。大部分患者平均视力低于0.1,且色觉严重缺失[1]。相对于患者较差的视力,瞳孔对光反射保留,且单眼发病时患侧眼相对性传入性瞳孔障碍并不明显。此现象发生机制可能与健侧眼亦发生视神经损害有关。另外一个假说为Leber患者视网膜中与瞳孔对光反射相关的视黑素细胞相对保留有关[5]。
眼底表现为该病最具特征性的临床表现之一。急性期视乳头充血、色红,毛细血管扩张、迂曲,视盘边界貌似模糊,极易误诊为急性视神经炎。Smith等[6]结合病理改变将典型LHON视乳头特征总结为3点:①视乳头周围毛细血管扩张样微血管病变(telangiectatic microangiopathy);②视盘周围神经纤维层肿胀(假性水肿,pseudoedema);③荧光素眼底血管造影视盘无渗漏。上述“经典”LHON视乳头表现可以很大程度帮助我们快速确诊(图1)。亚急性期视盘充血逐渐消退,视乳头黄斑束丢失。慢性期视盘呈现弥漫性萎缩[7](图2)。中心视野受累,即中心暗点、旁中心暗点和连生理盲点的中心暗点,是LHON另一重要临床特征(图3)。虽然大多数LHON患者仅有视力受损,但部分患者可同时伴有其他神经系统损害,如运动障碍、肌张力异常、共济失调、癫痫、听力障碍及肌病等[7],临床称为Leber叠加综合征(Leber plus)。
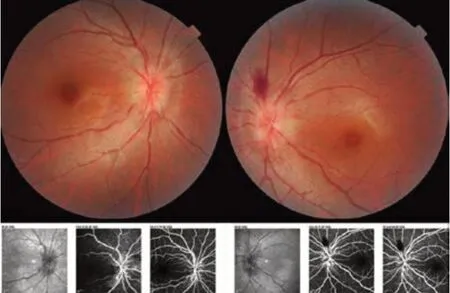
图1 LHON患者急性期双侧眼底 双侧视盘边界欠清,毛细血管扩张充血,左侧视盘鼻上方片状出血;下图荧光素眼底血管造影显示双眼均未见荧光渗漏,为假性水肿。
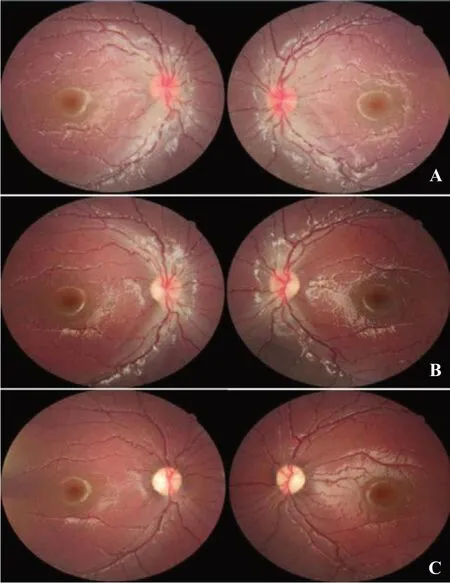
图2 LHON患儿发病不同时期眼底表现 A.急性期双眼视盘毛细血管扩张充血、色红、假性水肿;B.亚急性期视盘充血消退,视乳头黄斑束萎缩;C.慢性期视盘弥漫性萎缩、神经纤维层丢失。
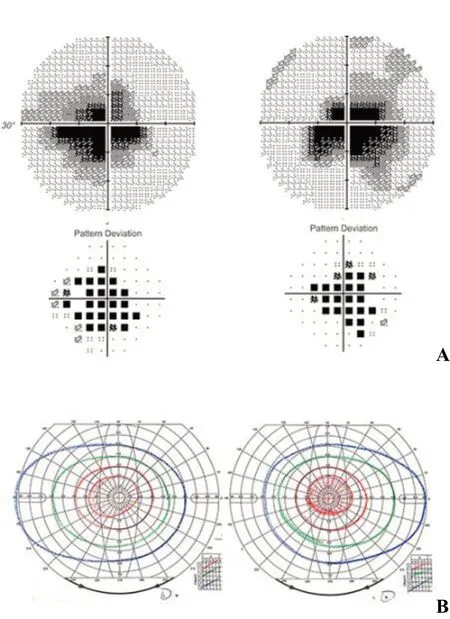
图3 LHON患者中心视野受累 A.Humphrey中心视野检查;B.Goldmann周边视野检查。
1.2 辅助检查 LHON的荧光素眼底血管造影显示视盘无渗漏,与检眼镜(眼底镜)下观察到的视盘肿胀、甚至盘周出血不同,因此LHON的视盘水肿实为假性视盘水肿。影像学检查通常具有鉴别价值:大多数LHON急性期双侧视神经无强化或轻度强化,借此与视神经炎进行鉴别。一些学者[8]发现LHON患者磁共振成像(magnetic resonance imaging,MRI)T2加权冠状位显示视交叉中心高信号,类似“面罩征”,是该病的另一个特征性表现。近年,随着光学相干层析成像(optical coherence tomography,OCT)技术广泛用于神经眼科领域,对不同时期患者视盘周围视网膜神经纤维层(retinal nerve fibre layer,RNFL)厚度和黄斑区神经节细胞-内丛状层(ganglion cell and inner plexiform layer,GCIPL)厚度2个角度进行评估,有助于预测疾病预后及鉴别诊断。急性期,由于视盘充血、毛细血管扩张,视乳头周围RNFL增厚;亚急性期和慢性期,视盘周围RNFL表现出颞侧变薄的趋势,为视乳头黄斑束损害导致(图4)。黄斑GCIPL在急性期即表现出明显变薄,不但有助于与视神经炎的鉴别,而且表明LHON是视网膜神经节细胞受累病变[9]。
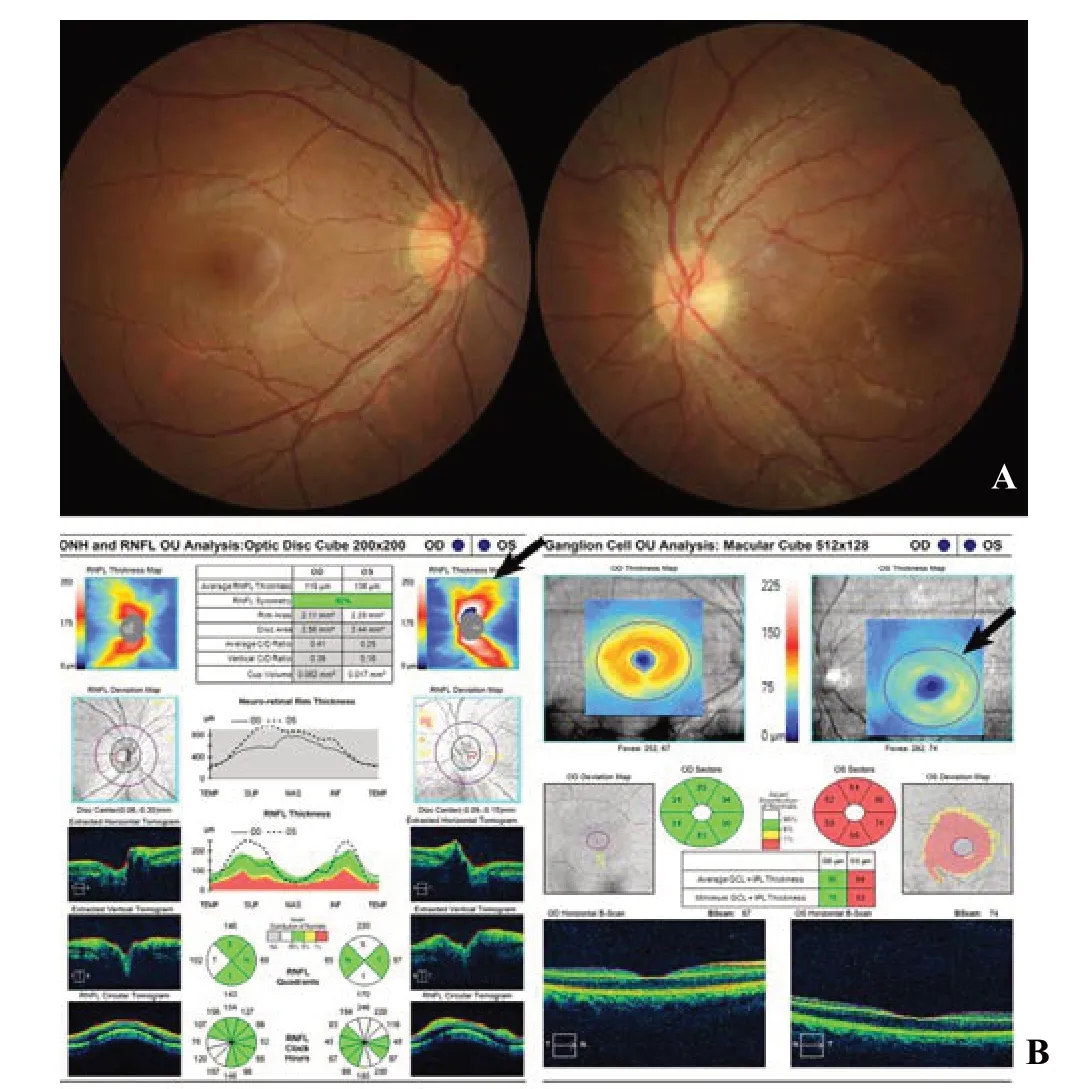
图4 LHON患者急性期眼底及OCT特征 A.患者左眼视力下降,左侧视盘边界欠清、颞侧色略淡;右眼虽无视力下降,但仍可见毛细血管扩张、充血的特征。B.Zeiss-Cirrus-OCT显示左眼视盘周围神经纤维层轻度增厚(左侧箭头),但黄斑节细胞明显变薄(右侧箭头),表明病变以神经节细胞损害为主。
1.3 基因诊断 国内基因检测技术最近10年发展非常迅速,从最初的聚合酶链反应(polymerase chain reaction,PCR)仅可检测3个原发突变位点的方式进入到目前对线粒体环基因全长的测序。因此,国内越来越多的继发位点突变导致的遗传性线粒体疾病被报道,且以往很多不明原因的视神经萎缩患者得以确诊[10]。尤其是在一些年龄偏大人群,很多为吸烟、酗酒及服用抗结核药物后诱发[11-12]。我们中心还报道了mtDNA12811点突变导致的眼肌麻痹患者[13]。目前,国内一些基因检测机构还可提供突变频率,有助于临床预测携带者的发病风险。本中心首诊眼科的视神经萎缩患者队列研究结果显示,249例遗传性视神经病变患者中LHON占67.9%[7]。精准的基因检测对于患者遗传生育具有重要的指导意义,且有望用于不久后的基因治疗。
1.4 治疗 目前尚无有效的药物治疗LHON,临床仍旧使用传统辅酶Q10及自由基清除剂艾地苯醌作为针对线粒体功能障碍的能量替代治疗。1项多中心、随机、双盲对照研究,即大剂量艾地苯醌(900 mg/d)治疗3个原发位点突变LHON临床研究显示:虽然治疗组与安慰剂组视力预后差异并无统计学意义,但该药物对于基线期双眼视力有差异患者的较好侧眼有改善作用[14]。2020年1项开放、多中心、回顾性、非对照Leber研究[15]显示,受试者最佳恢复的平均幅度为23个字母,观察终点时增加到36个字母(7行ETDRS);且在最后1次评估中,与最低点相比,视觉结果显著改善。目前,国内大剂量艾地苯醌治疗LHON的临床三期研究正在展开中。近年来,人们关注的LHON患者基因治疗也取得了初步结果:RESCUE、REVERSE及后续随访RESTORE研究均为腺相关病毒(adeno-associated virus,AAV)AAV转染ND4(rAAV2/2-ND4)治疗mtDNA11778突变LHON的疗效及安全性分析[16-17]。其中RESCUE纳入病程小于半年的LHON患者;REVERSE纳入了病程6~12个月的患者;RESTOR研究则是前述2个三期临床研究患者长达5年的随访,尚在进行中。初步研究结果显示,该基因治疗相对安全,未见严重不良事件。患者最佳矫正视力(best corrected visual acuity,BCVA)从病程12个月平均1.57logMAR(约小数视力0.03)提高至病程48个月时的1.26 logMAR(约小数视力0.06)。虽然数据显示BCVA的提高尚不足以显著改善患者的生活方式,但不失为根治LHON研究方向的尝试。
国内基因治疗虽然掀起了一股热潮,但要获得真正突破性的成果尚需较长时日。针对儿童患者低视力训练、视觉辅助设施及相关教育机构的完善可以很大程度提高患者的生活质量。健康宣教,包括戒烟、戒酒以及避免肝毒性药物的过度使用可以一定程度避免携带者发病。
1.5 预后 在3个原发突变DNA(mtDNA)11778、14484和3460中,14484患者预后最佳,视力甚至可恢复至1.0;11778患者预后不佳[1,4]。仅4%的11778突变患者可自愈;而14484的自愈率高达65%。
2 常染色体显性遗传性视神经病变
常染色体显性遗传性视神经病变(autosomal dominant hereditary optic atrophy,DOA)是 除LHON以外另一大类线粒体遗传性视神经病变。DOA虽然可伴有眼外症状,但核心损害为神经节细胞内能量代谢异常[2]。目前发现导致DOA的是OPA1~OPA8基因突变,其中65%~90%的DOA为OPA1基因突变导致[18]。
2.1 临床特征 DOA是国外文献中报道最常见的遗传性视神经病变,其发病率估计为1/50 000,在丹麦甚至高达1/10 000[19-20]。近期由于国内基因检测方法的进步,越来越多的OPA1基因突变导致的DOA患者得以确诊。患者发病年龄多在10岁之内,常见为4~6岁,尤其是在学龄前儿童体检筛查中被发现视力不佳及视盘苍白者居多。由于视力受损发病隐袭,呈现缓慢下降或稳定的趋势而非急性视力丧失,因此很多患者在成年后因“不明原因”的视神经萎缩而就诊。患眼视力在不同人群中差异很大,甚至同一家系中不同患者之间亦有不同。约40%的患者双眼视力可终身维持在0.3~1.0;视力为0.03~0.1的患者只占15%;严重的视力下降至光感或手动者少有[21]。眼底表现为视盘颞侧的楔形苍白萎缩,与视乳头黄斑束的神经纤维丢失相对应,易误诊为青光眼的杯盘比增大。在视野损害文献中,报道以中心视力及旁中心暗点居多,但临床中我们发现正常视野及旁中心视野缺损,类似双颞侧偏盲,尤其是双侧颞上的缺损类型,更为常见(图5)。推测与患者代偿性的旁中心注视有关。利用OCT技术观察黄斑节细胞丢失与视野损害的对应关系有助于鉴别[22]。我们中心研究显示,DOA患者年龄分布呈现双峰型:<10岁患者及35岁患者常见。推测后者为幼时发病但因视功能尚好未及时就诊。除视力损害外,DOA患者常伴有的其他神经系统症状为眼球震颤、斜视、眼外肌麻痹及神经性耳聋[7]。
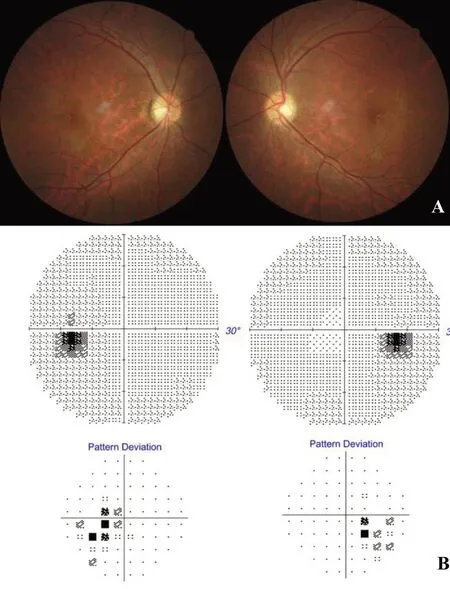
图5 DOA(OPA1基因突变)患者眼底及视野 男性32岁,体检时发现双眼视神经萎缩,双眼BCVA为0.3。A.眼底示双侧视盘小、苍白,尤其以颞侧楔形萎缩为著,神经纤维层萎缩;B.Humphrey视野显示双眼颞侧视野缺损,与LHON患者中心暗点不同。
2.2 基因检测OPA1基因为核基因,位于核染色体3q28–29。其编码的蛋白质为类动力蛋白GTP酶,是线粒体内膜融合及网状结构功能维持的重要蛋白;还参与了线粒体氧化磷酸化复合物及呼吸链膜蛋白的稳定。因此,OPA1基因突变导致的蛋白功能缺失最终引发线粒体破碎而出现神经节细胞凋亡[23]。目前已发现超过200个OPA1基因突变位点,但一些突变的临床意义尚待明确。这些突变包括蛋白质翻译提前终止、错义突变、移框突变以及重复大片段缺失等。借助PCR技术检测未发现突变的患者,使用基因二代测序后发现很多为OPA1基因大片段缺失或重排导致病变。因此,OPA1基因突变具有临床多样化的表现。除视神经萎缩外,内耳、脑、周围神经可伴发损害,即DOA叠加综合征(DOA plus syndrome)。
3 Wolfram 综合征
Wolfram综合征临床特征为青少年患I型糖尿病、进展性视神经萎缩、病程中不同时期出现尿崩症及神经性耳聋。因此简称为DIDMOAD(尿崩、糖尿病、视神经萎缩、耳聋的英文首字母缩写)。糖尿病症状多出现在10~20岁,并伴随着进展性视神经萎缩。少数患者以视力下降伴随视神经萎缩为首发症状。尽管视盘已呈现严重萎缩,但发病初期视力下降可不明显;随后逐渐进展,晚期视力多低于0.1。尿崩症和听力下降因人而异,出现于病程不同时期,听力下降首先累及高频听力。其他神经系统异常包括共济失调、癫痫、震颤、胃肠道功能紊乱、眼睑下垂、白内障及眼肌麻痹等(图6)。该综合征中位生存年龄为30岁,常见死亡原因为中枢性呼吸衰竭。本中心1例患者死于糖尿病酮症酸中毒。
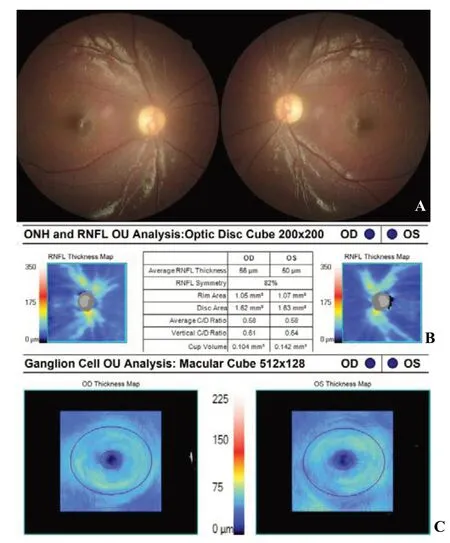
图6 Wolfram综合征患者眼底及OCT 女性6岁,自幼双眼视力不佳、先天性白内障,I型糖尿病。A.眼底示双侧视盘弥漫性苍白、萎缩,伴晶状体混浊;B和C.Zeiss-Cirrus-OCT示视盘周围神经纤维层(B)及黄斑神经节细胞层(C)弥漫性丢失。
大多数导致Wolfram综合征的基因WFS1位于染色体4p16.1。隐性遗传的方式常见,但偶有显性遗传方式。少数患者基因突变位于染色体4q22–24的CISD2(WFS2)基因,临床偶见[24]。WFS1基因编码蛋白与线粒体、内质网钙离子内流相关,因此Wolfram综合征也归属于线粒体疾病谱系。
4 线粒体脑病
Leigh综合征也称为亚急性坏死性脑炎,因突变基因不同其遗传方式可为常染色体隐性遗传、X-连锁或母系遗传的方式。症状通常出现在2岁以内,患儿呈进行性脑干功能障碍、共济失调、癫痫发作及周围神经病变,智力减退伴听力及视力下降。NDUFS1、NDUFS4、NDUFS7、NDUFS8、NDUFV1突变均为线粒体复合体Ⅰ功能异常,SDHA为复合体Ⅱ功能异常。除此之外,很多影响线粒体呼吸链组装、蛋白转运及功能维持的相关基因突变均能导致Leigh综合征或Leigh相关病变。临床表现除上述经典中枢神经系统病变外,尚可累及心脏、肝脏、肾脏、骨骼肌及周围神经、耳蜗等器官[25-26](图7)。视神经萎缩也可见于下述线粒体综合征,尽管视神经萎缩为临床次要表现:线粒体脑病伴乳酸血症及类卒中发作(MELAS)、肌阵挛癫痫伴破碎红纤维(MERRF)、慢性进行性眼外肌麻痹(CPEO)及Kearns-Sayre综合征等。我们的队列研究中也显示以LHON为唯一症状的mtDNA8344突变的MERRF综合征患者以及mtDNA13515突变的MELAS综合征患者。
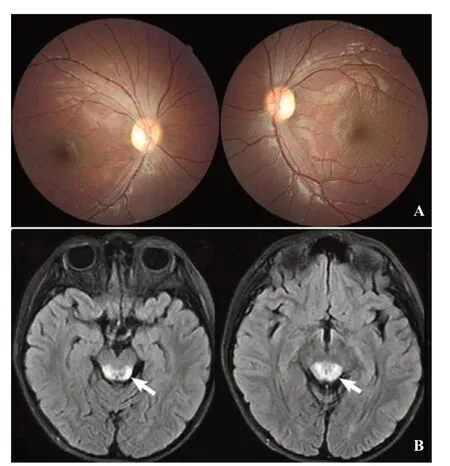
图7 Leigh综合征患儿眼底及颅脑MRI A.眼底示双侧视盘颞侧苍白,双眼球外旋(患儿有眼球运动障碍);B.颅脑MRI T2Flair示中脑背侧异常高信号(箭头)。
5 其他
Charcot-Marie-Tooth(CMT)病、遗传性痉挛性截瘫(hereditary spastic paraplegia,HSP)、Friedreich 共济失调及脊髓小脑共济失调(spinocerebellar ataxias,SCA)等均为伴有视神经萎缩的线粒体相关疾病。其中CMT病是神经系统最常见的遗传性周围神经病变,由MFN2(1p36.2)基因突变导致。MFN2编码线粒体外膜类动力蛋白GTP酶区域,作为OPA1蛋白结构与功能的补充,对线粒体氧化磷酸化起着重大作用。因此,该病视神经萎缩的临床特征与OPA1导致的病变类似[27]。由SPG7(16q24.3)基因突变导致其编码蛋白paraplegin功能障碍的疾病被确认为常染色体隐性HSP的一种临床类型。双眼视神经萎缩为该类患者典型的临床特征。SPG7与OPA1水解后2个片段的功能活化密切相关。Friedreich 共济失调为GAA三核苷酸重复导致的常染色体隐性遗传性疾病。FXN(9q13-21.1)基因编码的frataxin蛋白直接参与线粒体内膜铁-硫聚集功能,因此突变后表现出伴有视神经萎缩的线粒体功能障碍性疾病[28]。SCA为一组临床以小脑脊髓共济失调为特征的遗传性中枢神经系统病变,目前诊断亚型已经多达20余种。很多亚型与CAG三核苷酸重复有关。患者视力下降不仅与视神经萎缩、眼球震颤有关,尚有部分类型合并视网膜病变导致的视功能下降。
6 结语
遗传性视神经病变是神经眼科传入障碍性疾病中导致视力下降、视神经萎缩的常见疾病,其中线粒体疾病所占比例较高。视神经萎缩可作为疾病的唯一症状(例如LHON),也可伴随其他神经系统或周围神经病变(例如DOA),部分患者具有多系统损害的综合征样表现。对于患者的确诊应从不同疾病的典型临床表现、体征、辅助检查入手,然后锁定相关基因进行精准的基因检测。目前遗传性视神经疾病的治疗属于国际难题,但基因治疗已经在某些突变类型明确的单基因突变疾病中进行临床试验,不久的将来有望解决该临床问题。
志谢:感谢复旦大学附属眼耳鼻喉科医院生物样本库及迈基诺基因公司对本研究的支持。
