正常眼压性青光眼患者线粒体相关基因组变异研究△
2022-11-25乔云圣陈宇虹孙兴怀陈雪莉陈君毅
乔云圣 陈宇虹 孙兴怀 陈雪莉 陈君毅
(复旦大学附属眼耳鼻喉科医院眼科 上海 200031)
正常眼压性青光眼(normal tension glaucoma,NTG)的特点是在没有眼压升高(未经治疗眼压<21 mmHg,1 mmHg=0.133 kPa)以及房角开放的情况下出现青光眼性视神经萎缩和相应的视野缺损[1]。在中国人口中,NTG发病率约为1%,占所有原发性开角型青光眼(primary open-angle glaucoma, POAG)病例的70%[2]。考虑到NTG患者的眼压处于正常范围,研究者提出了其他病理生理学假说,包括血管功能障碍[3]、跨筛板压力梯度增加[4-5]、免疫疾病[6-7]以及遗传缺陷[8-9]等。
青光眼性视野改变是由于视网膜神经节细胞(retinal ganglion cell, RGC)的损伤和丢失。RGC发出的轴突直到筛板后才被髓鞘包绕[10],为了完成电信号的传递,需要线粒体提供大量能量。据估计,每个细胞都含有数百个线粒体,每个线粒体都有2~10套DNA拷贝,即线粒体DNA(mitochondrial DNA, mtDNA)[11]。mtDNA编码13种蛋白质(氧化磷酸化的重要亚单位)、22种tRNA和2种rRNA[12]。除mtDNA外,许多核基因也参与构建和维持线粒体的结构和功能[13]。
目前已有许多疾病确定为线粒体病,例如:Leber遗传性视神经病变[14]、线粒体脑肌病伴乳酸酸中毒和卒中样发作(MELAS)[15]和母系遗传的Leigh综合征[16]等。而OPA1(一种调节线粒体内膜融合的核基因)的变异可以引起常染色体显性遗传的视神经萎缩[17]。越来越多的研究提示,POAG与线粒体基因组突变以及单倍群之间可能存在联系[18-21]。在其他神经退行性疾病如阿尔茨海默病和帕金森病中,研究者也观察到由遗传因素引起的线粒体功能障碍[22]。
在本研究中,我们将核编码的线粒体基因和mtDNA基因结合起来定义为线粒体相关基因组(参考MitoCarta2.0[13]),并首次尝试通过高通量测序来探究其变异与NTG的相关性。
1 资料与方法
1.1 资料 本研究纳入了自2017年6月~2018年2月在本院治疗的10例NTG患者、10例高眼压性原发性开角型青光眼(high tension POAG, HTG)患者和10例年龄相关性白内障(age-related cataract,ARC)患者。所有患者均为汉族,彼此之间没有亲属关系。患者充分了解研究目的、取样方法、可能的风险和益处。每个参与者都签署了知情同意书。本研究获得了复旦大学附属眼耳鼻喉科医院人类研究伦理委员会的批准,并遵守《赫尔辛基宣言》的基本原则。
NTG诊断标准:存在青光眼性视神经病变,包括盘沿丢失和对应的视野缺损,房角开放,21 h眼压监测值正常(<21 mmHg)。HTG的诊断标准与NTG相同,只是眼压高于正常上限(>21 mmHg)。ARC组纳入标准:裂隙灯下观察到晶状体混浊,同时标准对数视力表检查视力≤0.5。排除标准:患有继发性青光眼(青光眼睫状体炎综合征、色素播散性青光眼、假性脱落综合征、葡萄膜炎等);患有其他视力和视野受损的眼科疾病(视网膜色素变性、糖尿病性视网膜病变、年龄相关性黄斑变性、病理性近视等)或有眼部表现的遗传性疾病。
1.2 方法 二代测序:使用High Pure PCR Template Preparation试剂盒(Roche,Basel,CH)从患者外周静脉血样本中提取基因组DNA。使用定制测序panel对37个mtDNA编码基因和1121个核编码线粒体基因[13]进行靶向富集与扩增。文库构建后使用Illumina Hiseq2500 测序平台 (Illumina,San Diego,CA,USA)进行高通量测序。使用Burrows–Wheeler Aligner (版本0.7.11)经过初步质控,读段与剑桥参考序列修订版(revised Cambridge Reference Sequence,rCRS)和人类基因组hg19进行比对。使用GATK(版本3.3)找到变异位点,使用ANNOVAR[23]对插入缺失位点(indels)和单核苷酸变异(single nucleotide variations,SNV)进行注释。
致病性预测:对于mtDNA变异,变异位点使用Mitomaster[24]进一步注释。符合以下标准的错义突变将被视为致病。①罕见变异:次等位基因频率(minor allele frequency,MAF)<0.5%(基于GenBank频率);②异质性>80%;③进化保守性>60%;④非同义突变;⑤未在对照组中出现;⑥文献报道为致病;⑦单倍群特异性。应用PON-mt-tRNA[25]、MitoTIP[26]和MSeqDR mvTool[27]来注释和评估tRNA突变;由于没有可用的注释工具,rRNA基因的变异通过查阅文献进行评估。
对于核基因变异,符合以下标准的变异被认为是致病的。①罕见变异:MAF(在国际千人基因组计划、gnomAD_genome、gnomAD_Exon、ESP6500、CG69和ExAC数据库中取最高值)<1%;②无义、移码、剪接位点和起始密码子突变;③在ClinVar数据库中被列为“致病”和“可能致病”的变异。符合以下标准的非同义突变将被纳入后续分析:①MAF<1%;②预测软件(SIFT、Polyphen2、PROVEAN、MutationTaster和MutationAssessor)中至少有4个给出有害预测;③GERP++得分>3,PhyloP 100way_vertebrate得分>1.5。
1.3 统计学处理 采用Stata 15.1和R 3.5.2进行统计分析。应用Fisher精确检验来比较3组之间的变异频率差异,采用Pearson卡方检验比较线粒体单倍群在各组间的分布差异。对于定量数据,采用Kruskal-Wallis检验,通过Mann-Whitney检验进行事后多重比较,并通过Bonferroni法调整校正P值。差异显著性阈值设定为0.05。
2 结果
2.1 mtDNA 变异分布 本次测序总共检测到296个变异(ARC组157个,NTG组130个,HTG组158个),详见图1。NTG组的转换(transition,Ts)与颠换(transversion,Tv)之比(Ts/Tv)以及非同义突变平均数量较高(表1),然而差异不具统计学意义。mtDNA基因间变异分布的比较:与ARC组相比,NTG组在12S rRNA基因中含有较少的罕见突变(NTG组0.1个/人,ARC组0.7个/人,P=0.009)。根据编码产物可将mtDNA分为几个功能区,即rRNA区(12S和16S)、复 合 体Ⅰ区(MT-ND1~6和MT-ND4L)、复合体Ⅲ区(MT-CYB)、复合体Ⅳ区(MT-CO1~3)和ATP合成酶区(MT-ATP6和MTATP8),tRNA编码区因其突变少而被排除。与ARC组相比,NTG组在rRNA区域含有较少的罕见变异(NTG组1.1个/人,ARC组1.7个/人,P=0.009)。
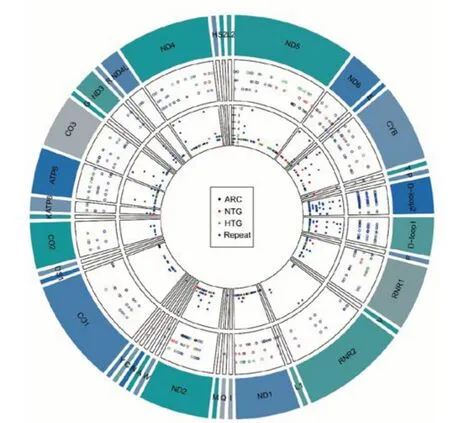
图 1 mtDNA变异分布 外圈表明了各个基因在mtDNA上的相对位置及大小。中圈的空心圆点标示了各个变异的位置。内圈的实心圆点指示了各变异的出现频次。其中黑色代表仅在ARC组出现的变异,红色代表仅在NTG组出现的变异,绿色代表仅在HTG组出现的变异,蓝色代表任意2组或3组中均存在的变异。
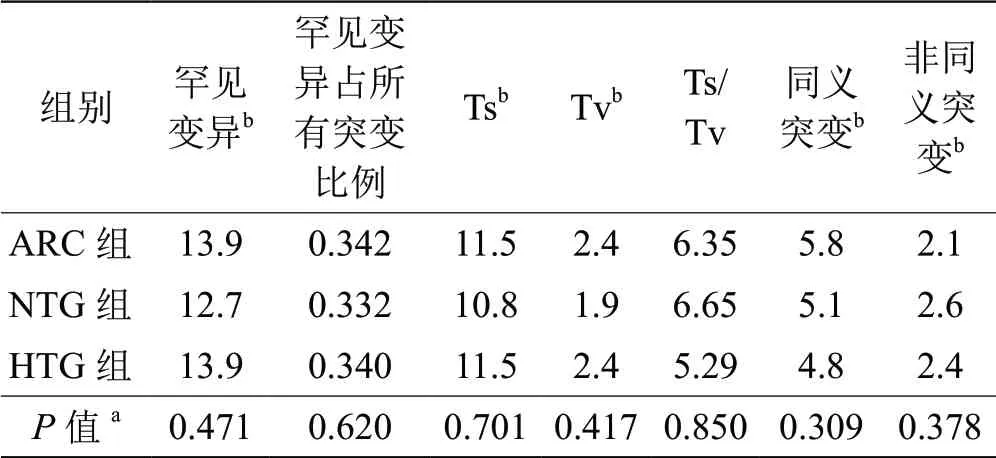
表 1 各类型罕见mtDNA突变平均数量在3组中的分布
2.2 线粒体单倍型分布 各组的单倍型分布列于表2。此患者群体共涉及8个亚型,包括M大类(D、M7)和N大类(H、B、A、F、R9、N9)。根据大类划分数据时,分布差异没有统计学意义(P=0.366)。
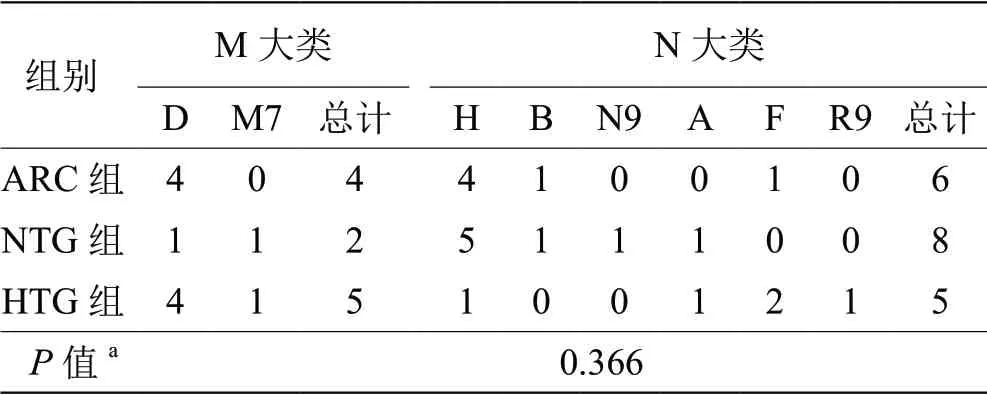
表 2 3组患者的线粒体单倍型分布(人)
2.3 线粒体相关核基因组变异分布 二代测序共检测到1 881个突变(ARC组1 276个,NTG组1 331个,HTG组1 214个),涉及754个基因(ARC组615个,NTG组641个,HTG组599个)。与ARC组相比,NTG组含有更多的缺失(NTG组8.5个/人,ARC组6.1个/人,P=0.006)和罕见的移码突变(NTG组1.6个/人,ARC组0.1个/人,P=0.002),与HTG组相比,NTG组的总突变数(NTG组557.7个/人,ARC组525.5个/人,P=0.005)和罕见突变数(NTG组34.8个/人,ARC组26.7个/人,P=0.002)更多,且罕见突变占总突变数比值更大(NTG组6.2%,ARC组5.1%,P=0.005)。
2.4 线粒体相关核基因组变异致病性分析 变异致病性评价的标准如“方法”一节所述。共有149个变异被认为是可能致病的(ARC组51个,NTG组59个,HTG组50个,P=0.491),涉及127个基因(ARC组45个,NTG组52个,HTG组47个,P=0.647)。虽然差异无统计意义,但是NTG组均为最高。
3 讨论
NTG缺乏青光眼的主要风险因素,即眼压升高。对潜在病理生理机制的探索有助于更好地理解疾病本质。在本研究中,我们推测遗传因素引起的线粒体功能障碍可能会导致NTG,因此对线粒体相关基因组进行测序,以探索可能的关联。
青光眼的根本病理改变是RGC损伤,表现为典型的视神经萎缩和视野缺损[10]。RGC发出的轴突构成视网膜神经纤维层,最终在视乳头处汇集[28]。RGC的轴突内含有大量线粒体[29],侧面反映了其高度的能量需求。三磷酸腺苷(ATP)的合成过程可产生大量活性氧,使mtDNA面临额外的损伤风险。事实上,mtDNA突变随着年龄的增长而累积[30],并被认为与年龄相关疾病和衰老本身相关[31]。综上所述,mtDNA突变符合NTG发病的时间性(老年)和空间性(RGC损伤)特征,提示mtDNA突变与NTG发病相关。
高通量测序技术的进步极大促进了遗传学研究。然而,测序偏差、生物信息大数据的处理等问题仍然阻碍了它在某些领域的应用。就mtDNA而言,其异质性[22]和镶嵌性[32]现象对测序技术的准确性提出了更高要求。Piotrowska-Nowak等[33]用Sanger测序和限制性内切酶片段长度多态性(RFLP)验证了二代测序检测到的每个mtDNA变异。他们还证明,二代测序技术可以检测到异质性低至5%的变异。在更高的测序深度下,可以捕获异质性只有1%的突变[34]。
已有许多研究报道了线粒体单倍群与青光眼之间的关系。在沙特阿拉伯人[35]和非裔美国人[20]中发现L单倍群与POAG有关,这与POAG在后者人口中的发病率较高相符[36]。另一方面,N1单倍群可能是高加索人种患青光眼的一个保护因素[35]。在波兰的一项关于NTG的线粒体基因组研究[33]中,与对照组(ARC)相比,H1单倍群在病例组中的所占比例较大。
在本研究中,NTG组在mtDNA中含有较少的变异,但在线粒体相关核基因组中含有更多的变异。例如我们观察到NTG组在12S rRNA以及rRNA区域的罕见变异比对照组少。Piotrowska-Nowak等[33]在其对照组中发现一些变异的频率较高。其中一些变异也在我们的样本中检测到,如:m.93A>G、m.310T>C、m.14470T>C,并且只出现在对照组。同样地,Singh等[18]报道,对照组中MT-ND2的罕见变异多于POAG患者。这些发现可能表明,罕见变异并不总是意味着较高的疾病风险,而可能为保护因素。另一项韩国的病例对照研究[37]在NTG患者中报道了3个与疾病相关的变异:m.4883C>T(MTND2:P138P)、m.9540T>C(MT-CO3:L112L)、m.14766C>T(MT-CYB:T7I),且m.4883C>T与 更 严重的视野缺损有关。然而,我们在研究中发现,前2个变异均匀分布在3组中,并且m.4883C>T是D单倍型的标记变异,在亚洲人群中比较常见。因此,对单倍型的忽视可能导致错误的相关性。
核基因组对线粒体功能的贡献甚至比mtDNA本身更大。MitoCarta[13]已经确定了一千多个线粒体相关基因。线粒体相关基因组的变化有可能会导致各种疾病。常染色体显性遗传的视神经萎缩(ADOA)是由OPA1基因突变引起的,OPA1编码一种线粒体内膜蛋白,调节线粒体的融合和分裂[38]。既往研究[39-40]表明,一些线粒体相关核基因与POAG和NTG有关,包括OPA1、MFN1、MFN2、PARL等。我们的研究也检测到了这些基因的变异。然而,在对突变频率、类型和组间分布进行过滤后,它们都不被认为是致病的。
综上所述,我们首次在中国患者中使用靶向二代测序技术探索NTG与线粒体相关基因组的关联,这为NTG发病机制的研究提供了有益参考。同时,我们也纳入了HTG患者,希望能在线粒体相关基因组方面区分这2种疾病。然而,我们的研究也存在一些不足:①研究群体较小;②从外周血样中提取DNA,不能反映RGC的实际情况;③没有用Sanger测序验证所有变异。但我们的初步探索能够提示NTG和线粒体遗传学之间存在关联。某些变异的存在可能是保护因素,当然这需要更大的队列和深入的试验来证实。
