基于遗传性眼病患者外显子测序数据的拷贝数变异检测工具的比较研究
2022-11-25兰远铮许晶乔王大利陈雪莉
兰远铮 许晶乔 王大利 陈雪莉
(1.复旦大学附属眼耳鼻喉科医院眼科 上海 200031; 2.复旦大学生命科学学院遗传工程国家重点实验室 上海 200438; 3.辽宁省大连市儿童医院中心实验室 大连 116012)
人类基因组广泛存在多种遗传变异形式。除了DNA的单个核苷酸突变,基因组还存在大片段DNA序列的变异。基因片段的拷贝数变异(copy number variants,CNV)是指一种序列长度为1 kb~3 Mb的DNA结构变异,通常包括单一片段的扩增、缺失[1]。CNV可以解释部分不由单核苷酸变异导致的人类遗传病和疾病易感性。CNV位点的突变频率比单核苷酸多态性(single nucleotide polymorphism,SNP)更高,是人类疾病的重要致病因素之一。CNV除了与单基因疾病及罕见疾病相关,也与许多人类复杂疾病如帕金森病[2]、阿尔茨海默病[3]、肿瘤[4]以及精神分裂症[5]相关。
全外显子组测序(whole-exome sequencing,WES)是一种广泛应用于临床与科研的二代测序技术,降低测序成本的同时提升了检测效能,它的出现推动了遗传病的研究进程。许多眼科疾病均与遗传相关,并表现出复杂的遗传学特性,一部分遵循孟德尔遗传定律,还有部分是由于多个基因的变异共同造成的。如青光眼就是一种遗传异质性的神经退行性疾病,目前有9个基因被人类孟德尔遗传数据库(OMIM)收录为青光眼的致病基因[6],但仍有大量的案例无法被已知的致病基因解释。其可能由WES中未发现的非编码变异、CNV等导致,这有待进一步深入研究。以原发性开角型青光眼(primary openangle glaucoma,POAG)为例,虽然在家系研究中发现大量基因位点与POAG呈现出基因-表型的相关性,但只有MYOC、OPTN、CYP1B1三个基因的单核苷酸突变和TBK1的CNV被明确证明存在于不同种族家系中[6]。此外,由于青光眼的遗传基因存在地区差异性,目前研究所得的基因不能广泛适用,并不能代表普遍现象[7-11]。因此,POAG的遗传学研究仍具有广阔的前景。针对与POAG类似的遗传性眼病,应考虑到其复杂的遗传机制、特异的基因地域分布性等因素。在WES技术日趋成熟的基础上,遗传眼病的研究应将重心从SNP扩大到CNV的分析上,以期为更多的患者确定致病基因,扩大各类遗传性眼病的突变谱。
在这项研究中,我们选取了CNV模拟生成的数据、公共数据库中的WES和panel的测序数据并进行分析,在4种适用于WES数据的CNV检测方法和软件中进行比较,旨在评估出合适的数据分析工具用于检测CNV,有助于遗传性眼病中CNV类型的致病突变分析的开展。
1 资料与方法
1.1 检测数据
1.1.1 模拟数据 我们同样用CNV模拟生成器VarSimLab[12]生成了模拟数据,用来评估CNV检测软件。VarSimLab是一款高度优化的模拟软件,输入FASTA格式的参考基因组和BED格式的目标测序片段(如WES中的外显子区域)后软件可根据参数生成一系列扩增或缺失的CNV区域。生成的文件包括:①一个列表文件,其中包括以txt格式保存的基准CNV的已知列表;②一个FASTQ文件,保存不含CNV的短序列作为对照组;③一个FASTQ文件,保存含有生成CNV的短序列样本。模拟数据中基准CNV的已知列表被用作计算精确度、灵敏度、错误发现率(false discovery rate,FDR)的金标准。
1.1.2 遗传性眼病患者的外显子组测序数据 我们从美国国立生物技术信息中心(National Center for Biotechnology Information,NCBI)的SRA数据库中下载了2组含有144个眼病基因panel的测序数据和1组遗传性眼病患者-正常配对的WES数据用来评估CNV检测软件。这些数据分别使用Illuminated HiSeq 2000平台测得。
1.2 检测WES数据的CNV工具 利用新一代测序技术(next generation sequencing,NGS)检测CNV的策略主要有4种:①基于读深(read count);②基于双端比对(paired-end);③基于序列组装(assembly);④基于分裂读段(split-read)[13]。基于读深的分析策略是NGS最常用的拷贝数检测方法,因为它的优点是适用性广。不管采用何种测序方法(单端或双端)都可以用此种方法分析。在本研究中,我们选用了最新发布且被广泛应用的4种基于读深分析策略的检测工具(表1):①CODEX;②CONTRA;③cn.MOPS;④VarScan2。

表 1 CNV检测工具
1.3 比较方法及指标 在本研究中,我们以灵敏度(sensitivity)、FDR、特异度(specificity)作为评价CNV检测软件的指标。以ratios(CNV值的比值)作为扩增及缺失的标准:若ratios> 检测阈值(Threshold=0.5)为扩增;若ratios< -Threshold为缺失;若ratios介于(-Threshold与Threshold)之间则认为没有CNV存在。
我们分别对每一种工具计算了灵敏度和FDR。按表2、3规则分别定义扩增和缺失情况下的真阳性(ture positive,TP)、假阳性(false positive,FP)、真阴性(ture negative,TN)和假阴性(false negative,FN),从而可以计算出灵敏度和FDR。
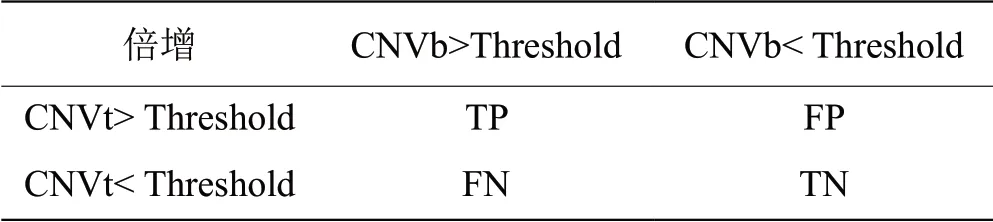
表 2 计算倍增情况下的TP、FP、TN和FN
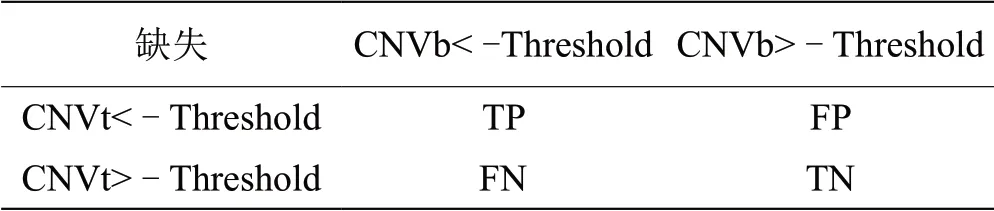
表 3 计算缺失情况下的TP、FP、TN和FN
2 结果
2.1 模拟数据结果 实验通过模拟生成器VarSimLab生成了一组测序深度为1 M的WES数据,用每个软件分别检测CNV。计算每种工具检测扩增和缺失的灵敏度和FDR(表4)。总体来看,每个工具都有良好的灵敏度(70%~90%)和较低的FDR(30%左右),且都能更好地检测扩增的情况。cn.MOPS(灵敏度88.70%)和VarScan2(灵敏度80.26%)分别在检测扩增和缺失时有良好的表现。cn.MOPS虽然在算法上降低了FDR且在检测扩增时稍优于其他软件,但是在检测缺失情况时的效果相对较差。
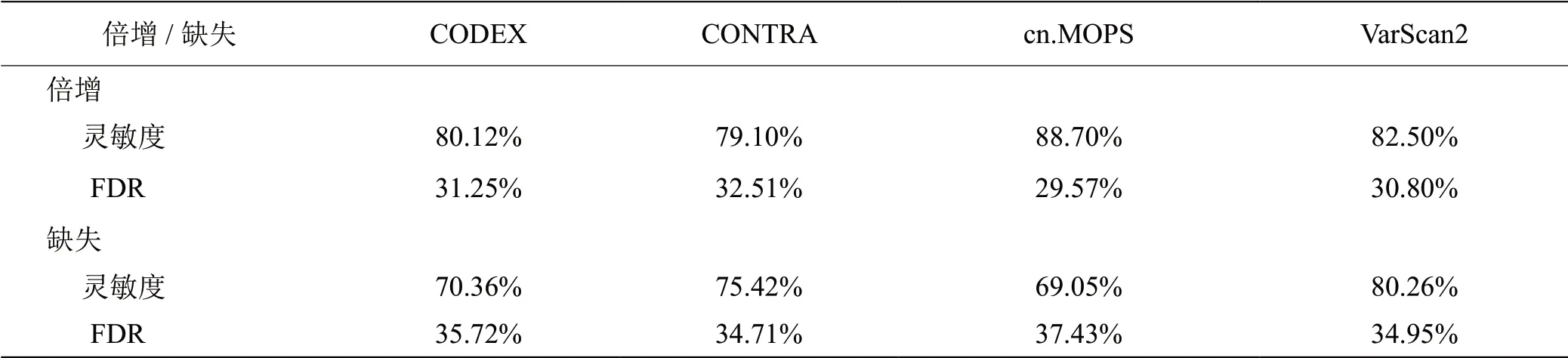
表 4 评估结果
2.2 WES测序数据与panel测序数据结果 为了进一步证实模拟数据的评估结果,实验采用了2组含有144个眼病基因panel 的测序数据(SRR1165160和SRR1165161)和1组遗传性眼病患者-正常配对的WES数据(SRR524806),通过其检测出的CNV数目来验证(表5)。WES检验结果基本与模拟数据的实验结果一致,但是在眼遗传病相关的panel测序数据中并没有检测到CNV。
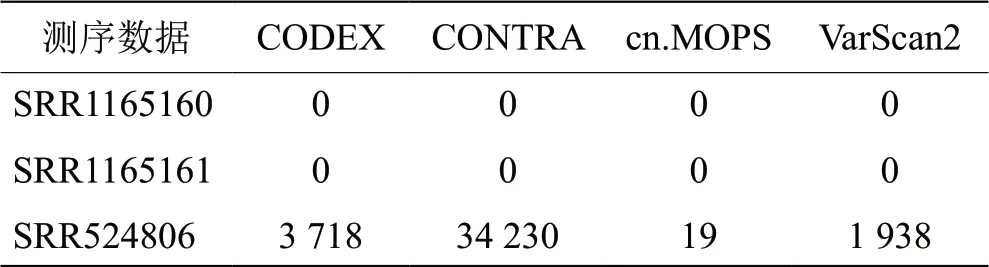
表 5 各软件检测出的CNV数目
2.3 软件运行速度 软件运行的速度也是我们在选择处理大量WES工具时需要考虑的重要因素。本实验记录了每个工具在处理同样数据时所耗费的平均时间(图1)。cn.MOPS的处理速度明显快于其他软件,CODEX和VarScan2平均耗时相对较长。
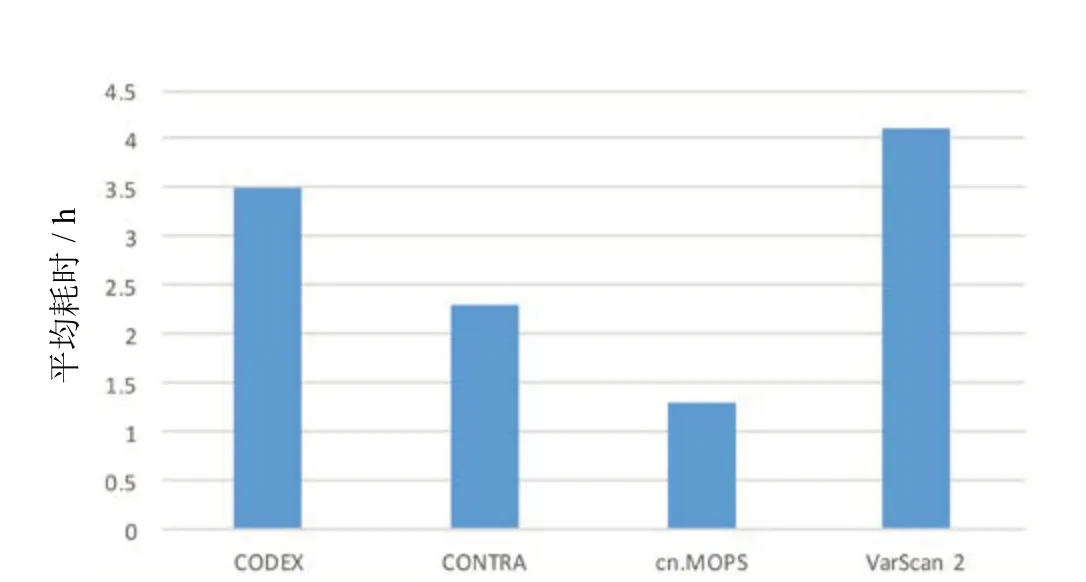
图1 每个工具的平均耗时 测试条件:基于Intel Xeon CPU E5-2650v2 @2.6GHz 1CPU 1M。
3 讨论
随着NGS[14]的迅猛发展,其高通量、高分辨率的优势,能够提供更精确和全面的基因组信息,基于高通量测序技术的CNV检测方法被更为广泛地应用[15-16]。全基因组测序(whole-genome sequencing,WGS)和WES是2种主要的NGS用于CNV检测的方法。WES是通过序列捕获将全基因组外显子区域(长度<2%基因组)捕获并富集后进行高通量测序的分析方法,具有对常见及罕见变异灵敏度高,测序序列短,测序成本较低的优点[17]。外显子区域是基因组中高度功能化的区域,在外显子区域的CNV相对于不在外显子区域的CNV更有可能成为致病基因。通过WES研究CNV成为了鉴定符合孟德尔遗传定律疾病致病基因的有效方法,也被用于复杂疾病易感基因的研究和临床诊断。
尽管自从NGS被用于CNV检测以来,开发出了很多方法和工具,但是很少有软件可以用于WES数据的CNV检测。现在已经开发了许多用WGS数据检测CNV的软件。但是这些软件要求所测得的数据连续分布;同时,用于WGS数据的软件并没有考虑WES由于捕获富集过程产生的偏差,所以这些软件并不适用于由WES检测出的数据[18]。除此之外,在此次panel数据的模拟分析中未检测到相关的CNV,其可能是因为panel的测序片段包含的基因比较少,且缺少合适的对照数据。目前所有可用于检测NGS目标(targeted)panel数据中的计算方法都存在一些不足[19]。而在现阶段,与WGS和panel相比,WES以其低测序成本和较全面的功能基因检测广度的优势,在临床与科研中的应用范围更广,具有更丰富的基因组资源背景。故研发出适用于WES的数据分析工具成为眼遗传学发展的迫切需求。
CODEX[20]通过同样的测序流程标准化样本后可实现多样本处理,其中标准化流程包括消除由GC含量、外显子长度、扩增效率引起的偏差以及潜在的人为引起的系统性偏差。CODEX还包括了一个基于柏松分布的递归分割程序,准确地模拟了基于计数的外显子测序模型。
CONTRA[21]是用于检测靶向重测序序列的软件,它可以用于检测小片段(100~200 bp)内的CNV。CONTRA软件均一化测序读段和对每个碱基取对数有利于均一化GC含量和低覆盖率区域的对数比计算。可以很好地解决GC含量过高或过低的覆盖区域测序读段相对减少的问题[22]。CONTRA很好地解决了二代测序本身覆盖率低和测序偏差问题,且适合读段不连续的全外显子测序数据。高FDR意味着很多检测出的CNV并不是真实存在的CNV,这会对后续分析产生很大的影响。
cn.MOPS[23]软件则通过用多个样本来判断最真实准确的CNV,可以很好地降低FDR。cn.MOPS合理地应用了贝叶斯法则原理,可以有效降低FDR。
VarScan2[24]是专门为了检验WES数据中的CNV开发的,尤其是癌细胞中CNV的检测。VarScan2可以同时分析肿瘤样本及其相应正常样本的序列数据,检测拷贝数变化。
针对此次测试的4种数据分析工具而言,cn.MOPS的平均耗时远小于其他三者,是耗时最长的VarScan2的30.49%。因此,针对大样本的人群基因组数据,在检测时选择cn.MOPS可显著提高基因分析的效率,有助于不同地区人群基因组学的研究及遗传流行病学研究;也适用于发病率相对较高的遗传性眼病研究,有利于人群中常见的致病性CNV的确定,或可进一步推动遗传性眼病早期基因筛查及干预研究的开展。并且cn.MOPS对于扩增突变具有更好的检测灵敏度和低FDR,对于CNV类型以扩增为主的遗传性眼病,cn.MOPS是理想的分析工具。而以扩增与缺失混杂或缺失为主的遗传性眼病,应选择cn.MOPS与其他分析工具相结合的方法,弥补cn.MOPS对缺失突变检测能力较弱的缺陷。
用于检测CNV的每个软件都有自己的优点和缺点,研究者可以根据需要选择合适的CNV检测软件。同时,软件开发人员可以参考每个软件的算法特点联合使用,也可以根据疾病相关基因的特点进一步开发相应的软件。
志谢:感谢上海昂朴生物科技有限公司对本研究提供方法学建议。
