肾移植术后脂蛋白肾病复发
2022-11-22董建华倪雪峰文吉秋葛永纯
董建华 倪雪峰 文吉秋 葛永纯
病史摘要
40岁男性患者,因“肾移植术后5年,尿检异常2月”,于2021-04-20入院。
现病史患者于2004年8月出现双下肢水肿,外院查尿蛋白定量5.5~12.2 g/d,尿红细胞10~15/HP,血清肌酐(SCr)67.5 μmol/L,血清白蛋白19.2 g/L,三酰甘油(TG)3.04 mmol/L,总胆固醇(CH)7.25 mmol/L,低密度脂蛋白胆固醇(LDL-C)4.16 mmol/L,肾活检示光镜下毛细血管腔内充满大块淡染栓塞物质,可见细胞性新月体、混合性新月体和纤维性新月体;免疫荧光提示载脂蛋白E(ApoE)+++~++++,诊断为“脂蛋白肾病(LPG)”,予泼尼松60 mg/d联合贝那普利治疗,尿蛋白未缓解(尿蛋白定量3~12 g/d),泼尼松逐渐减量至停用。2007~2009年期间服用中药汤剂偏方。2010年11月检查发现SCr升高,外院行重复肾活检仍提示LPG,光镜下见球性硬化比例62.8%,见细胞纤维性新月体,予甲泼尼龙冲击(500 mg×3 d),继泼尼松30 mg/d+雷公藤多苷90 mg/d治疗,尿蛋白无明显减少,后泼尼松逐渐减量,间断服用瑞舒伐他汀降脂,尿激酶抗凝,尿蛋白未减少,SCr逐渐升高。2016年6月SCr升至1 041.4 μmol/L,开始规律血液透析治疗。
2016年9月在外院行右侧同种异体肾移植术(DCD供肾,HLA两错配),术后SCr降至正常,尿蛋白阴性,泼尼松+他克莫司+吗替麦考酚酯免疫抑制治疗,依血药浓度调整免疫抑制剂用量。2021年2月复查尿蛋白0.46 g/d,SCr正常,外院移植肾活检光镜下可见1个毛细血管袢腔内见栓塞样物质,免疫荧光示ApoE++,诊断考虑移植肾LPG复发。2021年4月15日我科门诊复查尿蛋白+,尿红细胞阴性,SCr 97.2 μmol/L,为进一步诊治入院。
既往史2016年5月行左前臂动静脉内瘘手术;2016年9月行右侧同种异体肾移植术;无外伤史、输血史和过敏史。
家族史患者母亲和父亲非近亲结婚,尿检均阴性。家族中无同样疾病患者。个人史和婚育史无特殊。
体格检查血压125/86 mmHg,心率90次/min,体重67 kg,身高169 cm,体质量指数23.6 kg/m2;心、肺查体阴性。右下腹见长约12 cm手术瘢痕,可触及移植肾,大小约11 cm×5 cm,质中,无压痛,边界清,未闻及血管杂音。左前臂动静脉内瘘区未触及震颤、未闻及吹风样杂音。双下肢无水肿。
实验室检查
血液 白细胞7 800/μL,血红蛋白156 g/L,血小板17.1万/μL。凝血酶原时间10.8 s,活化部分凝血活酶时间24.0 s,纤维蛋白原1.63 g/L,国际标准化比值0.94。血清白蛋白36.0 g/L,球蛋白14.6 g/L,血钾3.56 mmol/L,血钠141.8 mmol/L,血氯108.1 mmol/L,总二氧化碳25.9 mmol/L,血磷0.7 mmol/L、血钙2.21 mmol/L,SCr 89.3 μmol/L,血尿酸305 μmol/L,谷丙转氨酶17 U/L,谷草转氨酶12 U/L,总胆红素16.2 μmol/L,TG1.13 mmol/L,CH 4.4 mmol/L,LDL-C 1.31 mmol/L,高密度脂蛋白胆固醇(HDL-C)1.37 mmol/L,ApoE 116.6 mg/L,ApoAl 1.53 g/L、ApoB 0.63 g/L,空腹血糖4.37 mmol/L,C反应蛋白<0.5 mg/L,估算的肾小球滤过率(eGFR)(CKD-EPI公式)90 mL/(min·1.73 m2)。
免疫学 免疫球蛋白IgG 6.41 g/L、IgM 0.427 g/L、IgA 1.18 g/L、IgE<17.1 IU/mL。淋巴细胞免疫分型CD20+117/μL、CD4+1 069/μL、CD8+577/μL。补体C3 0.81 g/L、C4 0.128 g/L。血κ轻链10.0 mg/L、λ轻链 14.3 mg/L,κ/λ 0.7;免疫固定电泳未见单克隆免疫球蛋白条带。群体反应性抗体Ⅰ类抗原A11为1 225,Ⅱ类抗原阴性。抗脐静脉内皮细胞抗体、抗磷脂酶A2受体抗体阴性。
尿液 尿蛋白定量0.74 g/d,尿红细胞0.4/HP,尿NAG 10.7 U/(g·Cr),尿RBP 0.4 mg/L。
辅助检查心电图:心率81次/min,窦性心律。胸部CT:双肺下叶少许纤维索条。移植肾血管超声造影:移植肾119 mm×58 mm×48 mm,移植肾肾主动脉及肾内各级血管树显示清晰,频谱多普勒显示波形正常,血管阻力指数正常,移植肾灌注未见异常。
外院肾活检
自体肾肾活检(2004-09-16) 光镜:23个肾小球,肾小球体积增大,毛细血管腔内充满大块淡染的“栓塞”物质,呈层状改变,毛细血管袢高度扩张,部分小球系膜细胞增生、系膜区增宽,部分节段可见泡沫细胞。可见球囊黏连及细胞性新月体、混合性新月体、纤维性新月体形成。大片状肾小管萎缩及间质纤维化,可见蛋白管型,多灶性炎性细胞浸润,为单核细胞及淋巴细胞。部分小动脉壁增厚,管腔狭窄。免疫荧光:系膜区及毛细血管袢节段性IgM+,毛细血管袢腔内栓子ApoE+++~++++。诊断LPG。
自体肾重复肾活检(2010-11-02) 光镜:43个肾小球中27个球性硬化,余肾小球呈弥漫性中-重度系膜细胞增生及系膜基质增多,可见局灶节段性硬化及球囊黏连,肾小球内见大块嗜伊红物质,7个小球见细胞纤维性新月体。多灶性肾小管萎缩、肾间质纤维化,灶性单核细胞及淋巴细胞浸润。小动脉壁纤维性增厚,部分小叶间动脉见透明变性。免疫荧光:IgM+、C3+。诊断LPG。
移植肾肾活检(2021-03-15) 光镜:15个肾小球中3个肾小球球性硬化,1个肾小球节段硬化;1个肾小球毛细血管袢扩张,可见栓塞样物质填充。肾小管上皮细胞轻度空泡变性,少数肾小管管腔扩张,刷状缘脱落;小灶状肾小管萎缩。肾间质少量淋巴细胞浸润、小灶状纤维化。肾小动脉内膜纤维性增厚。免疫荧光:毛细血管袢腔内ApoE、ApoA和ApoB阳性。电镜:肾小球基膜节段性增厚,足突节段性融合,节段性足突剥脱,基膜裸露,个别系膜区见少量低密度电子致密物沉积。诊断移植肾可疑LPG。
基因测序检查患者及其父母进行全外显子基因检测显示,患者ApoE基因型为ε3/ε3型,患者及其父亲ApoE基因第4外显子部分缺失(c.480-488delCAAGCTGCG,p.L162-R163-K164del),患者及其母亲肌动蛋白结合蛋白(ANLN)基因变异(c.115C>T,p.R39X),均为杂合变异。根据美国医学遗传学与基因组学学会指南,致病性分析显示ApoE(p.L162-K164del)初步判定为意义未明,ANLNR39X为疑似致病性变异,可能致局灶节段性肾小球硬化(FSGS),但文献数据库无两个位点变异报道,ClinVar数据库无上述位点致病性分析结果。对基因变异区域进行Sanger测序验证,结果见图1和图2。

图1 ApoE基因变异区域Sanger测序验证结果(↓所示为基因变异起始位置)

图2 ANLN基因变异区域Sanger测序验证结果(↓所示为基因变异位点)
诊疗分析
本例患者临床特点如下(1)中年男性;(2)基础疾病为LPG;(3)肾移植术后泼尼松+他克莫司+吗替麦考酚酯免疫抑制治疗,SCr降至正常,尿蛋白阴性;(4)肾移植术后5年,肾损害表现为少量蛋白尿,移植肾病理为肾小球毛细血管袢脂蛋白栓子形成,ApoE染色阳性;(5)血ApoE明显升高;(6)ApoE(p.L162-K164del)基因突变。结合患者临床、肾病病理及基因检测结果,初步诊断为移植肾LPG复发。
患者肾脏病理同时出现毛细血管袢脂蛋白栓子和肾小球泡沫细胞浸润的非典型LPG表现,需与以下ApoE相关肾小球疾病相鉴别:(1)ApoE2纯合子肾小球病:临床表现为不同程度蛋白尿,伴高脂血症和血ApoE升高,特征性病理表现为肾小球泡沫细胞浸润。而在ApoE2杂合ApoE Tokyo和ApoE Maebashi突变时,病理可同时表现脂蛋白栓子和非层状泡沫细胞浸润,但本例患者ApoE基因类型为ε3/ε3型,非ε2/ε2基因型。(2)膜性肾病样载脂蛋白E沉积病:为ApoE2/2+ApoE Toyonnaka(Ser197Cys)基因突变导致ApoE在肾小球基膜上皮侧沉积所致,可表现为脂蛋白血栓和泡沫细胞,但病理可见基膜“钉突”形成,本例患者基因类型和病理均不符合。
患者携带ANLNR39X突变基因,病理可见肾小球局灶节段硬化、足突融合,但LPG也可有此病理表现,患者是否存在遗传性FSGS目前不能确定。
最终诊断(1) 移植肾LPG复发;(2) 右侧同种异体肾移植术后。
治疗2021-04-28~2021-05-28予离心/膜分离组合式双重血浆置换(DFPP)清除ApoE治疗,共9次。同时予非诺贝特0.2 g/d控制血脂,泼尼松20 mg/d、吗替麦考酚酯0.5 g 2次/d、他克莫司1 mg 2次/d、雷公藤多苷片20 mg 3次/d抑制免疫,缬沙坦80 mg/d及对症治疗。
DFPP采用动静脉穿刺作为血管通路(图3),全血流经连续血浆分离装置Cobe Spectra(Cobe Laboratories, Lakewood, CO)进行血浆分离,血流量55 mL/min,分离机转速2 400 r/min。抗凝剂为4%枸橼酸液,与血液的输入速度比为1∶20。血浆分离速度25 ml/min,分离出的血浆进入二级滤器血浆成分分离器EC50W(Asahi Kasei Medical CO., Ltd.日本),滤出血浆回输至患者体内。单次DFPP处理血浆量为5 358±212 mL,弃浆685±173 mL。DFPP治疗后血ApoE下降59.6±25.7 mg/L,IgG下降2.1±0.3 g/L,纤维蛋白原下降0.71±0.19 g/L,检测废液ApoE浓度115.8±21.2 mg/L(含盐水),单次DFPP治疗ApoE清除量约为71.8 mg。治疗过程中患者未发生过敏、低血钙、出血及感染等并发症。
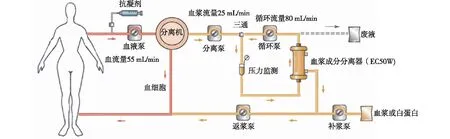
图3 离心/膜分离组合式双重血浆置换示意图
2021-05-31行移植肾重复活检:光镜下28.6%(10/35)球性废弃,余肾小球体积增大,系膜区轻度增宽;25.7%(9/35)肾小球节段血管袢高度扩张,袢腔内充满PAS、HE淡染层状栓子,内皮细胞成对。肾小管间质轻度慢性病变,小灶性小管萎缩、基膜增厚,少量小管炎,间质小灶性单个核细胞浸润。小动脉节段透明变性。免疫荧光:IgM+,呈颗粒状弥漫分布于系膜区;IgG、IgA、C3、C1q阴性;袢腔内容物ApoA+、ApoB++,ApoE阴性。电镜:肾小球系膜区、内皮下和上皮下未见电子致密物沉积,足细胞足突融合<5%(图4)。
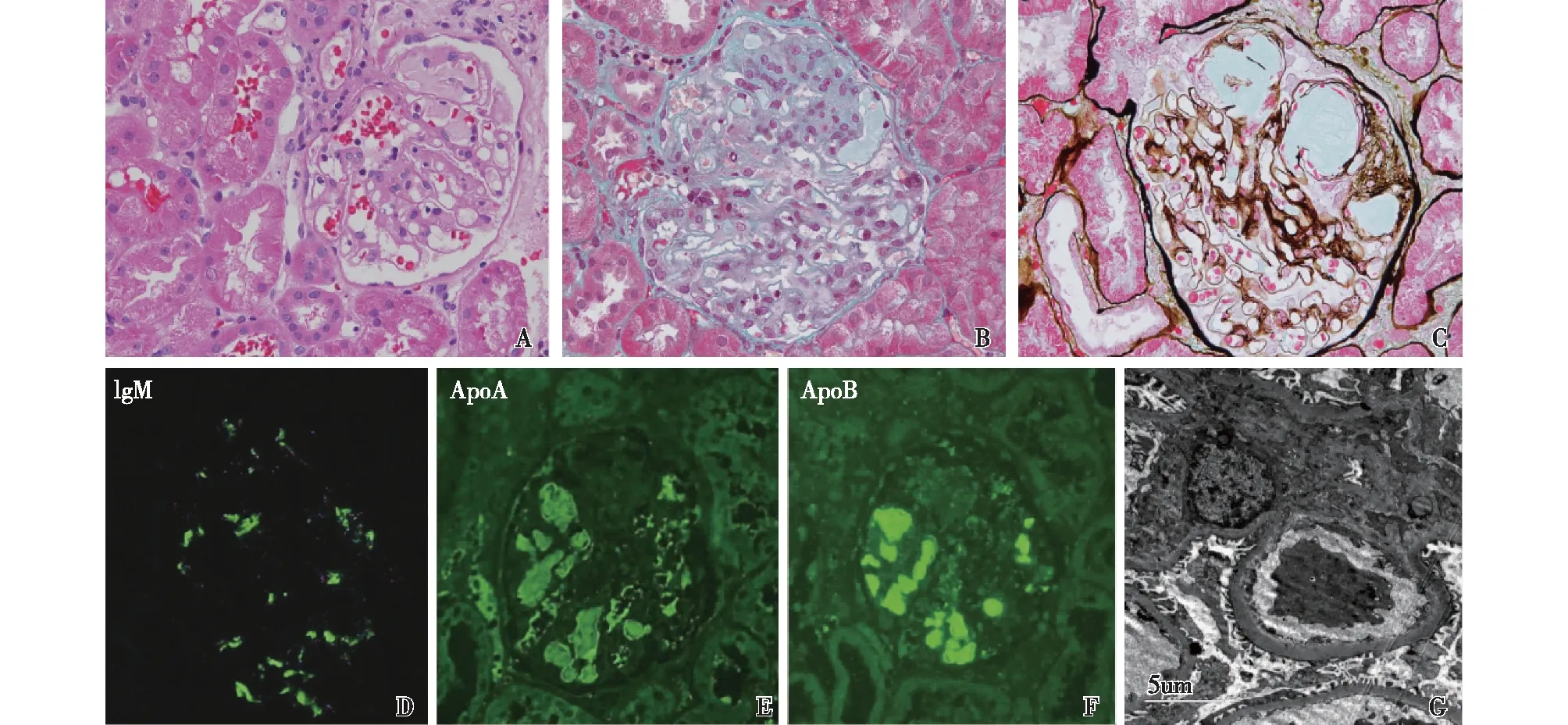
图4 移植肾病理A,B,C:肾小球毛细血管袢内充满淡染、不嗜银、层状结构物质(A:HE,×400;B:Masson三色,×400;C:PASM-Masson,×400);D:lgM呈颗粒状沉积于系膜区;E:袢腔内容物ApoA阳性;F:袢腔内容物ApoB阳性(D、E和F:IF,×200);G:肾小球系膜区、内皮下和上皮下未见电子致密物沉积(EM)
2021-06-02复查尿蛋白定量0.42 g/d,SCr 114.0 μmol/L,ApoE 99.5 mg/L,ApoA 1.2 g/L,ApoB 0.39 g/L,TG 1.0 mmol/L,CH 3.1 mmol/L,LDL-C 0.71 mmol/L,HDL-C 1.27 mmol/L,群体反应性抗体Ⅰ类抗原A11为881。
讨 论
LPG是一类以蛋白尿、血ApoE升高,伴高脂血症为主要临床特征,肾小球毛细血管袢脂蛋白栓子形成为主要病理特点的一种罕见肾小球疾病。ApoE是由299个氨基酸组成的糖蛋白,分子量为34kD;N-端结构域含低密度脂蛋白受体(LDLR)结合区,C-端结构域含脂类结合区[1]。ApoE通过C-端结构域与脂类结合,成为乳糜微粒、极低密度脂蛋白、LDL-C和HDL-C的重要组成部分。上述脂质蛋白被脂蛋白脂肪酶分解后,脂蛋白残余颗粒上的ApoE可通过与肝细胞LDLR结合,使其被肝细胞摄取并代谢,从而降低血脂水平[2]。ApoE基因突变是LPG主要致病机制。人ApoE基因含有4个外显子和 3个内含子,具有3种等位基因(ε2、ε3和ε4),其中ApoE3/E3型最常见[3]。LPG相关的ApoE基因突变目前至少有20种,主要是LDLR结合位点或附近的基因缺失或错义突变。ApoE基因突变造成ApoE清除受限和高脂蛋白血症,是引起脂蛋白在肾小球毛细血管袢沉积的重要原因。
目前ApoE基因缺失致LPG的主要有三种类型:ApoE-Tokyo(p.Leul141-Lys143del)[4],ApoE Maebashi(p.Arg142-Leu144del)[5],以及ApoE(p.Gln156-Gly173del)[6]。文献报道的ANLN基因突变引起家族性FSGS的仅有ANLN(c.1852G>T: p.G618C)和ANLN(c.1291C>T: p.R431C)[7]。本例患者ApoE(p.L162-K164del)和ANLNR39X基因突变类型均属首次发现和报道,其致病机制暂不清楚,需在转录组学、蛋白质组学、拓扑学方面进一步研究以全面阐释其致病机制。ANLN突变基因通过诱导足细胞凋亡引起家族性FSGS,而本例患者肾脏病理见肾小球局灶节段硬化、足突融合,但LPG也可有此病理表现,其是否存在遗传性FSGS尚不能确定。本例患者同时携带两种导致肾脏疾病的突变基因,可能是引起新月体形成等非特异性肾脏病理表现的原因。
患者父亲为ApoE基因突变携带者,目前没有肾脏受累的临床表现,尿检未发现异常,血ApoE亦在正常范围。可见LPG发病受多因素影响,基因突变不是唯一因素。有部分患者临床和病理均为LPG典型表现,但ApoE基因测序显示外显子编码区序列与正常人一致,可能有非编码区变异[8]。非编码区调节编码蛋白的表达、剪切和修饰折叠。非编码区基因突变可能引起ApoE变构,表现为基因型和蛋白表型分离。任何引起脂蛋白受体识别、结合或者酶作用改变的因素均可能导致脂蛋白的异常代谢和在组织中沉积。
LPG临床尚缺乏有效的治疗手段,治疗以降低血脂,减少蛋白尿和延缓肾功能进展为主要目标。患者病初应用糖皮质激素、免疫抑制剂、血管紧张素转换酶抑制剂和抗凝治疗,目前证明上述治疗基本无效。非诺贝特和苯扎贝特可有效降低LPG患者TG,以及尿蛋白和血ApoE水平,延缓肾功能进展,而他汀类药物治疗效果不明显[9-10]。
国家肾脏疾病临床医学研究中心将双重血浆置换(DFPP)和免疫吸附应用于LGP治疗,并取得良好临床疗效。葡萄球菌蛋白A(SPA)免疫吸附治疗LPG中,SPA 通过其氨基端Fc段结合区与ApoE非特异性结合,从而有效清除血循环中ApoE。免疫吸附治疗后重复肾活检,显示肾小球毛细血管袢内脂蛋白栓子减少或消失;复发患者重复性免疫吸附治疗不仅能取得与首次治疗相同疗效,并使疾病缓解期延长[11]。但因ApoE免疫吸附为非特异性吸附,IgG下降明显,感染风险增加,限制了其在临床应用。DFPP治疗后LPG患者尿蛋白、SCr和血ApoE水平显著下降,部分患者接受10次DFPP治疗后肾组织脂蛋白栓子几乎完全消失,ApoE降至正常范围,短期疗效明确[12]。本例患者在接受DFPP治疗前后血ApoE下降明显,但停止治疗后,血ApoE仍会升高,因单次治疗仅能部分清除血循环中致病物质,同时沉积于组织的脂蛋白代谢释放ApoE。因此,建议DFPP应与贝特类药物联合治疗。DFPP治疗应在LGP疾病早期进行,对于肾小球硬化比例高、肾小管间质慢性化病变严重的患者效果不佳。
本例患者虽接受足疗程DFPP治疗,但移植肾重复活检显示脂蛋白栓子并未完全消失,免疫荧光ApoE染色却转为阴性,可能与以下因素有关:(1)首次移植肾活检可能未完全反映肾脏病理改变。在积极DFPP治疗期间,血ApoE水平下降,而肾小球毛细血管袢脂蛋白栓子反而短时间内快速形成,不符合疾病进程;(2)毛细血管袢内脂蛋白栓子未消失,可能与特殊类型的ApoE基因突变类型和巨噬细胞功能异常有关。ApoE基因突变造成极低密度脂蛋白清除障碍,是引起脂蛋白在毛细血管袢沉积的重要原因。ApoE-Kyoto+富含TG的脂蛋白与脐静脉内皮细胞结合能力较正常ApoE增加30%~50%[13]。ApoE-Chicago与肾小球毛细血管壁结合能力增强,可能是通过受体结合而非蛋白聚糖结合[14]。本例ApoE(p.L162-K164del)可能存在类似机制导致脂蛋白栓子与毛细血管袢结合紧密,不易被清除。巨噬细胞在ApoE相关肾脏疾病发生和发展中发挥重要作用。ApoE2纯合子相关肾小球病[15]和ApoE5相关肾脏病[16]中可见巨噬细胞源性泡沫细胞浸润,可能与巨噬细胞功能活化有关。相反,在LPG中却很少能观察到泡沫细胞浸润,大量脂蛋白在毛细血管袢腔沉积并形成栓子。动物模型研究证实[17],巨噬细胞表面清道夫受体CD36表达下降,导致LDL清除受限,可能也是影响脂蛋白栓子清除的重要因素。(3)毛细血管袢腔内脂蛋白栓子虽未消失,但脂蛋白内ApoE因浓度梯度差增大释放,导致ApoE染色阴性。这也可能与DFPP疗程不足,以及重复肾活检的时间相距DFPP治疗结束时间太短有关。
因LPG为ApoE基因异常疾病,LPG肾衰竭患者肾移植后结构和功能异常的ApoE仍持续产生,故目前报道的5例LPG肾移植患者,均在肾移植术后2年内复发,其中2例患者术后未降脂治疗,3年内移植肾失功[18]。本例患者在肾移植术后5年出现蛋白尿,肾脏病变出现应更早,故建议常规移植肾活检监测LPG复发情况。早期治疗干预可预防和延缓移植肾LPG复发的发生和进展[18]。
小结:本文首次报道1例LPG肾移植术后复发,并同时携带ApoE(p.L162-K164del)和ANLNR39X突变基因,DFPP联合非诺贝特治疗后,血ApoE水平下降。此类患者常规移植肾活检有助于监测移植肾LPG复发情况,以便早发现、早干预,以改善远期预后。
