绿豆RS4·Se(IV)制备、结构表征及对酶活抑制动力学
2022-10-31赵姝婷王维浩全志刚刘德志王一飞武云娇苏有韬魏春红曹龙奎
赵姝婷,王维浩,2,全志刚,王 娟,刘德志,王一飞,武云娇,苏有韬,魏春红,曹龙奎,2,3,
(1.黑龙江八一农垦大学食品学院,黑龙江 大庆 163319;2.国家杂粮工程技术研究中心,黑龙江 大庆 163319;3.黑龙江省天然产物模拟移动床色谱分离技术创新中心,黑龙江 大庆 163319)
绿豆原产于亚洲的印度东北部、缅甸等地,其中碳水化合物占63%,膳食纤维占16%,是淀粉的重要来源。绿豆淀粉因其高直链淀粉含量和高交联特性,可用于生产具有多种益生作用的抗性淀粉(resistant starch,RS)。RS在小肠中不消化,但是可以在大肠中发酵和吸收,对于调节血糖水平,改善糖脂吸收和促进肠道有益菌增值方面有着重要作用。RS分为5 种类型,其中化学改性淀粉被归类为RS4。RS4抗消化能力与取代基引起的消化酶攻击空间受阻有关,取代基的引入导致更大的立体位阻效应,阻止了酶-底物复合物的形成,使其具有抗降解性,如醚化、交联和酯化。目前柠檬酸酯化淀粉的制备方法主要为干热法、湿热法和电位循环法。当淀粉和柠檬酸混合体系受热时,酸攻击淀粉链,柠檬酸酐将淀粉链上的羟基取代。干热酯化法是被用于生产改良淀粉制剂、食品工业、制药等最常用的方法。添加其他化学成分的食品安全性难以控制,柠檬酸作为较安全的食品添加剂是近年来应用最广泛且安全的制备RS4的物质之一。
硒是动植物生长发育必需的微量元素,不能自身合成,只能从食品和硒补充剂中获得。硒在自然界中的存在形式可根据硒的形态分为有机硒和无机硒。与无机硒相比,有机硒更容易被人体吸收。硒多糖因其稳定性强、生物活性高被广泛应用于硒补剂中。由于天然硒多糖中硒含量较低,因此需要通过硒化获得硒含量丰富的有机硒多糖。硒多糖组成分为单聚糖和杂聚糖两类,主要连接方式为1-3糖苷键,绝大多数为-构型。硒化修饰是通过NaSeO与半缩醛羟基(C-OH)和酯基发生反应形成,利用糖链上的羟基、氨基等活性基团与硒化试剂中的含硒化合物通过共价键结合在糖链上,柠檬酸酯化淀粉因具有较多的半缩醛羟基和酯基可作为硒化的主要原料。有研究表明,平菇、苦瓜、龙骨等植物硒化多糖有显著降糖作用,可能是由于它们具有对-淀粉酶和-葡萄糖苷酶的抑制活性。王峙力等将甜玉米芯多糖进行硒化后发现对两种酶的抑制作用显著升高。同时,硒是谷胱甘肽过氧化物酶的重要组成部分,能促进受损胰岛细胞修复及葡萄糖代谢降低血糖水平,Lian Kexun等通过研究硒化甘草多糖体内抗氧化活性,发现血液和肝脏中谷胱甘肽过氧化物酶和超氧化物歧化酶的活性显著升高,肝肾中丙二醛的含量显著降低。RS4也可通过抑制与糖代谢有关酶活抵抗糖类消化,二者协同改善体内糖代谢情况。本研究以RS4为原料进行硒化,在抵抗消化的基础上增强对酶的抑制作用,同时硒的引入可以增强抗氧化活性,修复受损的胰岛细胞,进一步强化其生物学功能。硒化绿豆RS4(selenized mung bean resistant starch,MB-RS4·Se(IV))可作为抗氧化剂和酶抑制剂,研究其可能在体内发挥的作用,为寻找新型、高效、低毒的辅助降糖补硒食品及药物提供初步实验依据。
本实验以绿豆抗性淀粉(mung bean resistant starch,MB-RS4)为原料,采用硝酸-亚硒酸钠法制备MB-RS4·Se(IV),并对硒化前后MB-RS4采用扫描电镜、紫外光谱、傅里叶变换红外光谱、X射线衍射、凝胶渗透色谱、核磁共振(nuclear magnetic resonance,NMR)测定颗粒形貌、相对分子质量及结构变化;考察体外对-葡萄糖苷酶和-淀粉酶抑制作用并进行酶促反应动力学分析,为硒化绿豆抗性淀粉在功能性食品和天然药物的开发领域提供实验基础和理论依据。
1 材料与方法
1.1 材料与试剂
绿豆淀粉(食品级) 市售。
亚硒酸钠 天津市大茂化学试剂厂;-葡萄糖苷酶(水解酶类,酶活力40~80 U/mg)、猪胰-淀粉酶(水解酶类,酶活力50 U/mg) 美国Sigma-Aldrich公司;4-硝基苯基---吡喃葡萄糖苷(4-nitrophenyl--glucopyranoside,pNPG) 上海阿拉丁生化科技股份有限公司;可溶性淀粉 北京北化精细化学品有限公司;所有无机试剂均为分析纯,购自天津市大茂化学试剂厂。
1.2 仪器与设备
DGA-9080A电热恒温鼓风干燥箱 梅特勒-托利多仪器上海有限公司;真空冷冻干燥机 基因有限公司;透析袋(m为1 000 Da) 上海源叶生物科技有限公司;S-570扫描电子显微镜 日本日立公司;D/MAX2000V型X射线衍射仪 日本理学制造公司;T6系列紫外-可见分光光度计 北京普析通用仪器有限责任公司;4500傅里叶变换红外光谱仪 美国Thermo Nicolet公司;GPC-20A高效凝胶渗透色谱(high performance gel permeation chromatography,GPC)仪、RID-20示差折光检测器 日本岛津公司;AVANCE III 600M NMR仪 德国布鲁克公司;SPECTROstar Nano酶标仪 德国BMG LABTECH公司。
1.3 方法
1.3.1 MB-RS4的制备
根据李蒙娜的方法并作适当修改。将淀粉与柠檬酸按照干基比5∶2混合,加入适量蒸馏水搅拌均匀,用氢氧化钠调节混合体系pH 3.5左右,室温静置12 h,于40 ℃烘干。取出干燥样品粉碎过60 目筛,取筛下物于150 ℃酯化4 h,反应产物用无水乙醇洗涤3 次,37 ℃烘干至水分质量分数为8%,即得MB-RS4(RS4质量分数为83.72%)。
MB-RS4测定方法:准确称取1 g样品,加入5 mL-淀粉酶和葡萄糖苷酶混合液后,取不同时间的水解液采用GOPOD法进行测定。
根据下式计算样品中RS4含量:
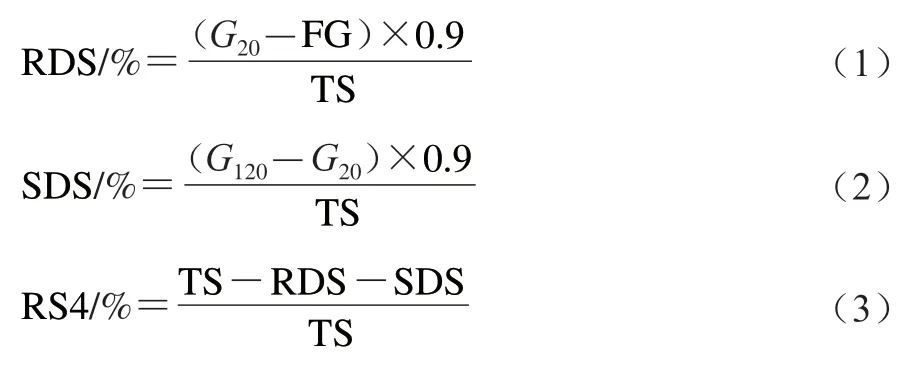
式中:和分别为淀粉酶水解反应20 min和120 min后产生的葡萄糖含量;TS为样品中的总淀粉含量/mg;0.9为换算系数。
1.3.2 MB-RS4·Se(IV)的制备
根据王丽波等的方法并作适当修改。用50 mL体积分数为0.5%的HNO溶液溶解样品(1.0 g),在室温下搅拌30 min,加入1.0 g NaSeO和0.7 g BaCl,将混合物在66 ℃搅拌反应3.5 h。冷却至室温滴加1 mol/L NaOH溶液调节pH值至7~8。加入0.5 g NaSO除去溶液中的杂质Ba,4 000 r/min离心15 min,取上清液后透析出反应产生的无机盐和多余的NaSeO、浓缩、醇沉,冻干即得MB-RS4·Se(IV),得率为26.450 6%。
1.3.3 MB-RS4·Se(IV)得率计算
按式(4)计算MB-RS4·Se(IV)得率:

式中:为MB-RS4·Se(IV)的质量/g;为MB-RS4的质量/g。
1.3.4 MB-RS4·Se(IV)中硒含量的测定
1.3.4.1 硒标准曲线的绘制
根据国标选用分光光度法测MB-RS4·Se(IV)中硒含量。测得硒标准曲线方程为=19.380 21-0.042 04,=0.995 7,其中:为硒含量/mg,为吸光度。
1.3.4.2 MB-RS4·Se(IV)中硒含量的测定
在酸性条件下MB-RS4·Se(IV)中Se可以与邻苯二胺生成络合物,络合物在330~340 nm处有紫外吸收特征峰,根据此原理测得MB-RS4·Se(IV)中的硒含量。
5 mg样品中加入2 mL HNO溶液(质量分数3%),4 ℃放置过夜。将样品加热至烧杯中无橙黄色烟雾生成,冷却至室温加入6 mol/L的盐酸8 mL继续加热至无白色烟雾生成,转移至25 mL容量瓶,加入2 mL邻苯二胺(质量分数2%)溶液,然后用HCl溶液定容并调节pH 2,避光反应1 h,最后用5 mL甲苯萃取,在334 nm波长处测定有机相吸光度,按下式计算样品中硒含量:

式中:为有机相中硒的质量浓度/(mg/mL);为定容后待测溶液体积(25 mL);为有机相总体积(5 mL);为待测溶液体积(2 mL);为MBRS4·Se(IV)质量(5 mg)。
本实验制备所得MB-RS4·Se(IV)中硒含量为2.21 mg/g。
1.3.5 MB-RS4·Se(IV)的结构测定
1.3.5.1 扫描电子显微镜观察
根据Asad等的方法并作适当修改对样品的形貌和微观结构进行研究。将样品置于双面胶带喷金,于50.0 kV收集图像。
1.3.5.2 紫外光谱测定
配制质量浓度为2 mg/mL样品溶液,蒸馏水作空白对照,注射器吸取待测样品,针孔过滤器过滤,190~400 nm波长范围内扫描,扫描间隔1 nm。
1.3.5.3 傅里叶变换红外光谱测定
根据Castrocampos等的方法并作适当修改对两种物质进行分析,将2 mg样品与200 mg溴化钾(KBr)粉末混合并压片于4 000~400 cm测定。
1.3.5.4 X射线衍射图谱分析
根据González等的方法并作适当修改。将干燥细腻的样品均匀分散于板框内压实,使得样品表面光滑平整,将样品框固定后测试。衍射测试条件:管电流40 mA、管电压40 kV、Cu靶波长1.540 6 Å、Co靶波长1.790 26 Å、扫描速率7°/min、2测量范围5°~70°。
1.3.5.5 GPC法测定相对分子质量及分布
采用窄分布聚乙二醇(polyethylene glycol,PEO)作标准曲线相对校正法。标准样品:窄分布PEO标样组;RID-20示差折光检测器;样品处理:取50 mg样品,加1 mL 90%二甲基亚砜溶液,溶解过夜,加2.5 mL无水乙醇,离心去除上清液,沉淀经无水乙醇洗涤2 次,风干后加入(0.1 mol/L NaNO+0.06% NaN溶液)溶解,于121 ℃反应20 min,5 000 r/min离心10 min,取20 μL上样检测;流速:0.6 mL/min,柱温:35 ℃。
采用TSKgel GMPWXL凝胶色谱柱分别对改性前后MB-RS4相对分子质量的测定。样品数据采用日本岛津HW-2000 GPC色谱工作站分析计算。
1.3.5.6 NMR测定
根据Li Min等的方法并作适当修改,将样品溶于DO,充分振荡使其完全溶解,D-NMR(H-NMR、C-NMR)。
1.3.6 MB-RS4·Se(IV)体外酶活抑制测定
根据Wang Hao等的方法并作适当修改。将-葡萄糖苷酶用0.1 mol/L(pH 6.8)磷酸盐缓冲溶液稀释成1 U/mL,配制pNPG浓度为5 mmol/L。测定时,向96 孔板加入50 μL样品溶液,同时加入50 μL pNPG溶液,37 ℃孵育10 min,取出加入100 μL-葡萄糖苷酶溶液37 ℃孵育45 min,加入50 μL浓度为0.2 mol/L的NaCO溶液终止反应,酶标仪测定405 nm波长处吸光度()。反应体系见表1,以阿卡波糖作为阳性对照,酶活抑制率计算按式(6)计算:
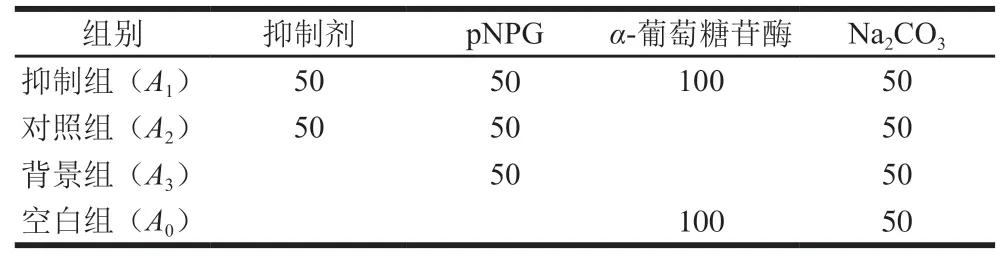
表1 α-葡萄糖苷酶抑制作用反应体系Table 1 Composition of reaction system for α-glucosidase inhibition μL

式中:为添加样品和酶时测定抑制组吸光度;为将酶液换为等体积缓冲溶液的对照组吸光度;为将样品替换为等体积ddHO的背景组吸光度;为将样品以等体积ddHO替代,酶液以缓冲溶液替代测定空白组吸光度。
1.3.6.2 抑制-葡萄糖苷酶动力学分析
固定酶浓度为1 U/mL,96 孔板中加入50 μL不同浓度样品溶液,同时加入50 μL不同浓度pNPG溶液,在37 ℃孵育10 min,取出加入100 μL-葡萄糖苷酶溶液37 ℃孵育45 min,加入50 μL浓度为0.2 mol/L的NaCO溶液终止反应,酶标仪测定405 nm吸光度。最大酶促反应速率()、米氏常数()等动力学参数可通过Lineweaver-Burk双倒数作图确定,即酶反应速率()的倒数1/与底物浓度(pNPG)的倒数1/[]的关系图。
1.3.6.3 对-淀粉酶的抑制作用
根据Sangilimuthu等的方法并作适当修改。用缓冲溶液配制浓度为2 U/mL的猪胰-淀粉酶和1%的可溶性淀粉,将不同浓度梯度的样品溶液40 μL与-淀粉酶40 μL混合37 ℃孵育30 min,加入40 μL可溶性淀粉后继续孵育10 min,然后加入160 μL 3,5-二硝基水杨酸(3,5-dinitrosalicylic acid,DNS)保温5 min煮沸显色,稀释2 倍体积后取200 μL于96 孔板,在540 nm测定抑制组吸光度,同时做对照组、背景组和空白组,通过式(3)计算抑制率,体系内所加溶液体积如表2所示。

表2 α-淀粉酶抑制作用反应体系Table 2 Composition of reaction system for α-amylase inhibition μL
1.3.6.4 抑制-淀粉酶动力学分析
将党员后备培养、党员教育管理、村级事务管理等纳入对党组织书记考核,强化督导检查,对出现的问题及时诊断、及时对症下药。同时加强对基层党组织书记干事创业的考核,将致富带富能力当做重要考核指标。
根据曾傲琼的方法并作适当修改。固定酶浓度为2 U/mL,40 μL不同浓度样品溶液中加入40 μL-淀粉酶混匀,于37 ℃孵育30 min,取出加入不同浓度可溶性淀粉40 μL,于37 ℃孵育10 min,加入160 μL DNS保温5 min煮沸显色,稀释2 倍体积取200 μL于96 孔板,在540 nm波长处测定吸光度。最大酶促反应速度()、米氏常数()等动力学参数可通过Lineweaver-Burk双倒数作图确定,即酶反应速率()的倒数1/与底物浓度(pNPG)的倒数1/[]的关系图。
1.4 数据处理与分析

2 结果与分析
2.1 MB-RS4、MB-RS4·Se(IV)表观及结构分析
2.1.1 扫描电镜分析
在相同放大倍数下,由图1a、b可知,用柠檬酸处理淀粉后,酯化淀粉颗粒呈团块状,表面有局部腐蚀、裂缝和小的重叠层,这些可能是由酯化反应引起的,与高粱淀粉中用酸处理报道结果一致。在柠檬酸淀粉颗粒样品中能够看到环形颗粒状,是因为在酸溶液中颗粒肿胀,然后在淀粉芯有组织区域塌陷并可能与柠檬酸作用有关,导致颗粒形态改变。图中框内为由于柠檬酸作用淀粉颗粒之间的交联处。而图1c、d中MBRS4·Se(IV)淀粉颗粒完全塌陷并形成各种不规则形式碎片化,框内淀粉颗粒破碎并且表面粗糙呈多孔的蜂窝状,这与Gu Yangeng等研究一致,由于硒化反应过程中HNO与亚硒酸钠反应生成HSeO,HSeO电离产生Se与MB-RS4发生反应,改变了范德华力和分子间相互作用,造成颗粒破碎,分子质量变小。

图1 MB-RS4、MB-RS4·Se(IV)扫描电镜图Fig.1 SEM images of MB-RS4 and MB-RS4·Se (IV)
2.1.2 紫外光谱分析
如图2所示,NaSeO在225 nm有一个强的特征吸收峰,通过柠檬酸处理绿豆淀粉得到MB-RS4,然后再对MB-RS4进行硒化处理得到MB-RS4·Se(IV)。在紫外吸收光谱(190~400 nm)发生明显改变,MB-RS4最大吸收峰所对应波长为220 nm,最大吸光度为0.677,并在272 nm处产生肩峰,为酯化反应产生的酯基发生电子跃迁造成。MB-RS4·Se(IV)紫外扫描最大吸收峰对应波长=248 nm,最大吸光度为1.223,明显不同于MB-RS4和NaSeO,但呈现相似峰形,Se与MB-RS4发生相互作用导致结构发生变化,Se核外有空轨道,可接受MB-RS4酯基的孤对电子形成配位键,影响C=O的电子跃迁,同时Se的d轨道电子受到配位体的影响发生跃迁,导致红移和增色效应。

图2 MB-RS4、MB-RS4·Se(IV)紫外光谱图Fig.2 UV spectra of MB-RS4 and MB-RS4·Se (IV)
2.1.3 傅里叶变换红外光谱分析
由图3可知,在3 438.15 cm和2 932.45 cm处为O—H的伸缩振动峰和亚甲基的C—H伸缩振动峰,是典型的糖类物质的伸缩振动峰。1 000~1 200 cm处为C—O—C和C—O—H的特征峰,说明两种样品中有吡喃糖苷的存在。MB-RS4在1 745.23 cm和1 644.21 cm处分别为C=O和—CO—的伸缩振动特征峰,说明MB-RS4中存在酯化糖醛酸,1 000~800 cm波数范围内的峰为样品中的和-构型。
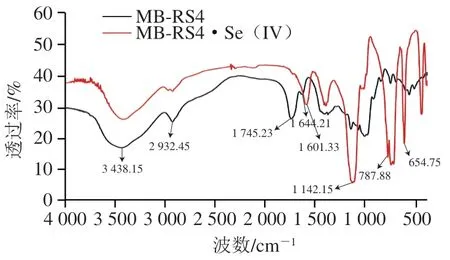
图3 MB-RS4、MB-RS4·Se(IV)红外光谱图Fig.3 Infrared spectra of MB-RS4 and MB-RS4·Se (IV)
MB-RS4·Se(IV)在1 745.23 cm处特征峰发生红移,说明酯化糖醛酸参与了硒化反应。1 000~800 cm波数范围内的峰在硒化后峰减少,说明构型发生了变化。MB-RS4·Se(IV)在787.88、730.88、654.75 cm处出现新振动峰。787.88 cm处为Se=O伸缩拉伸振动峰,说明反应体系中亚硒酸与酯基发生取代反应。730.88 cm为C—C(Se)伸缩振动峰,是由于在反应过程中糖环发生断裂形成不饱和键,游离Se在C=C上发生顺式取代造成。654.75 cm为Se—O—C的伸缩振动峰,是由于亚硒酸与C6上的羟基发生脱水反应形成。
2.1.4 X射线衍射图谱分析
李良玉、Kaur等研究发现绿豆淀粉X射线衍射图呈现C型结晶尖峰特征,结晶度较高。从图4可以看出,MB-RS4的结晶结构被破坏,趋于一条弥散曲线,直观看结晶度不高,2在18.38°左右,为V型结晶峰。说明柠檬酸处理后的MB-RS4淀粉颗粒轻度破坏,仍然存在部分结晶区;但是在MB-RS4·Se(IV)的衍射图中发现,MB-RS4·Se(IV)曲线呈现弥散状态,结晶结构被完全破坏,这与扫描电镜结果一致。

图4 MB-RS4、MB-RS4·Se(IV)X射线衍射图Fig.4 X-ray diffraction patterns of MB-RS4 and MB-RS4·Se (IV)
2.1.5 分子质量及分布
由图5可以看出,MB-RS4中出现2 个组分峰,MBRS4·Se(IV)中只有1 个组分峰(17 min后为容器或溶剂中的小分子杂质峰,故17 min后出现的峰不做分析)。在MB-RS4的图谱中可以看出第1个峰出现的保留时间在7.643 min,峰面积占比48.49%,分子质量为6.8×10Da;第2个峰出现的保留时间在16.403 min,峰面积占比33.74%,分子质量为4.869×10Da。从图5B可以看出,峰出现的保留时间在16.090 min,峰面积占比24.61%,分子质量为8.012×10Da;由图5可以看出,MB-RS4中6.8×10Da分子质量占比较大,而MBRS4·Se(IV)则是以8.012×10Da分子质量占比较大。Tan等利用凝胶色谱对绿豆淀粉分子质量进行测定,测定结果表示绿豆淀粉分子质量的分布范围在1.82×10Da和1.54×10Da之间。经由柠檬酸处理的绿豆淀粉发生酯化和脱水反应,将两个或两个以上的淀粉分子聚合,呈现稳定的多维空间网状结构,聚合后形成大分子的MB-RS4,分子质量显著增加。而在硒化过程中由于添加的硝酸和生成的亚硒酸又作用于MB-RS4,使MB-RS4分子链发生断裂,导致硒化后分子质量显著降低。

图5 MB-RS4(A)、MB-RS4·Se(IV)(B)凝胶渗透色谱图Fig.5 Gel permeation chromatograms of MB-RS4 (B) and MB-RS4·Se (IV) (B)
由图6 可以看出,M B-RS4 中存在独立的两个组分,分子质量分别在3.91×10~1.26×10Da和3.62×10~0.29×10Da的范围内。大分子质量段先出峰,小分子质量段后出峰,其中大分子质量物质在两种组分中占比较大,且呈现正态分布,而小分子质量组分则呈现偏正态分布;MB-RS4·Se(IV)中只存在一个组分,分子质量分布在9.04×10~0.2×10Da范围内,为小分子质量组分,硒化后的小分子质量段呈现正态分布,分布较均匀。MB-RS4中存在大量的酯化、交联后形成的大分子物质,所以MB-RS4中大分子质量段所占比例较大。而在MB-RS4·Se(IV)中由于发生了硒化反应,反应过程中酯基和糖苷键的断裂导致大分子质量组分转化为小分子质量组分,致使小分子质量段比例增加。
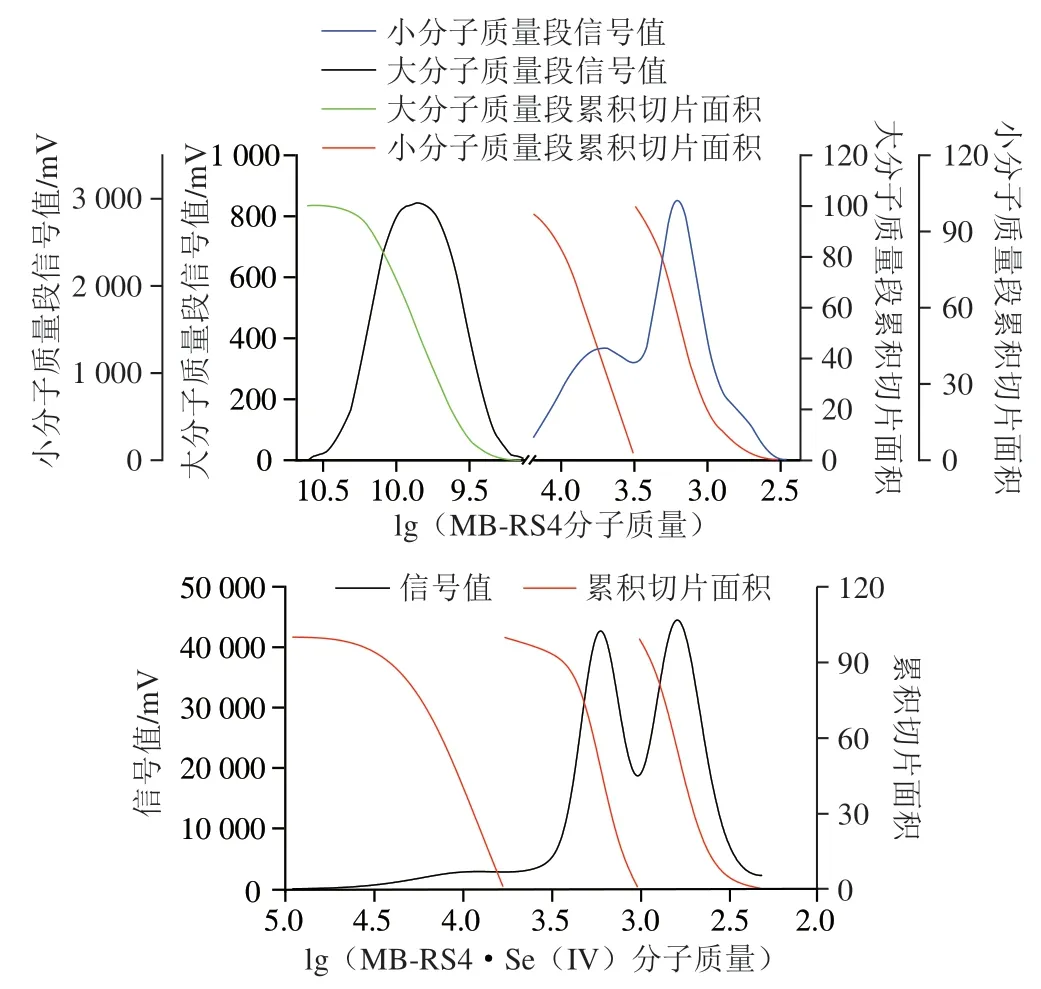
图6 MB-RS4与MB-RS4·Se(IV)相对分子质量分析图Fig.6 Relative molecular mass analysis of MB-RS4 and MB-RS4·Se (IV)
从表3 可以看出,MB-RS4 的数均分子质量m=2.26×10Da,重均分子质量m=1.88×10Da,分散系数为8.3×10;MB-RS4·Se(IV)的m=1.3×10Da,m=1.20×10Da,分散系数为9.22。由以上可以看出,MB-RS4的分散系数较大,分子质量分布范围较广,而硒化改性后分散系数显著降低,说明改性后体系更加均一,组成更简单。
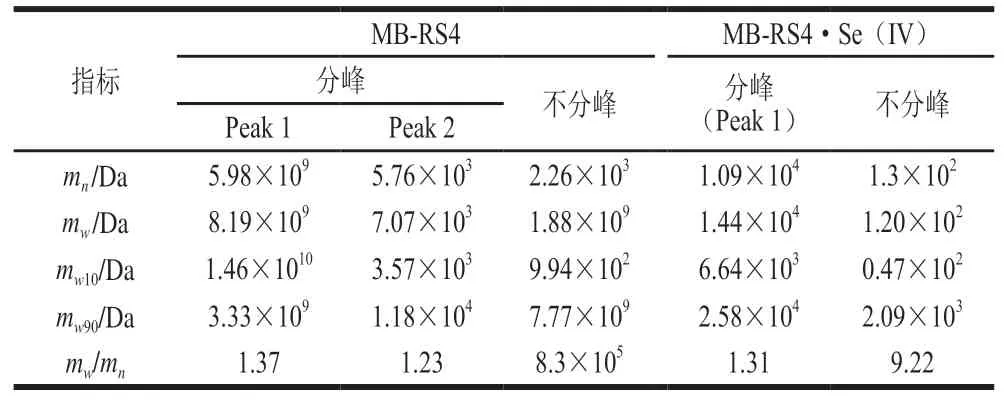
表3 MB-RS4与MB-RS4·Se(IV)相对分子质量测定结果Table 3 Relative molecular mass of MB-RS4 and MB-RS4·Se (IV)
2.1.6 NMR分析
MB-RS4·Se(IV)、MB-RS4 1H-NMR谱图见图7。图8中显示化学位移出现在4.98和1.35,一般情况下,化学位移大于5.00表现为糖类化合物的构型,化学位移小于5.00表现为糖类化合物的-构型。MB-RS4的异头氢化学位移出现在4.98,说明MB-RS4为-构型。C谱中,MB-RS4位于61.45处的信号峰归属于糖环中的C6信号峰,72.66处的信号峰归属于C2、C3,位于82.09处信号峰属于C4的信号峰,C4的强度与V型结构相关。MB-RS4中82.09中峰强度较大,说明MB-RS4主要以V型晶谱存在。同时,MB-RS4中102.00处出现一个单峰,可能来自相同范围信号强度的V型复合物的信号峰,这些现象共同验证了MB-RS4的X射线衍射图谱中18.38°处的峰为V型结晶峰。173.65处信号峰为MBRS4中柠檬酸酐与淀粉共振的碳酯峰。
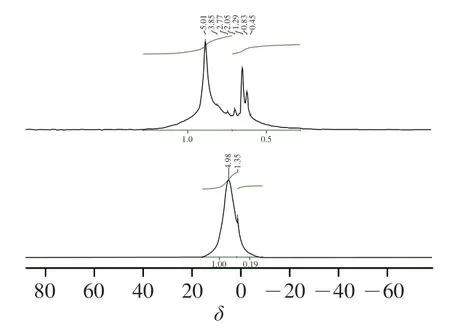
图7 MB-RS4·Se(IV)、MB-RS4 1H-NMR谱图Fig.7 1H-NMR spectra of MB-RS4·Se (IV) and MB-RS4
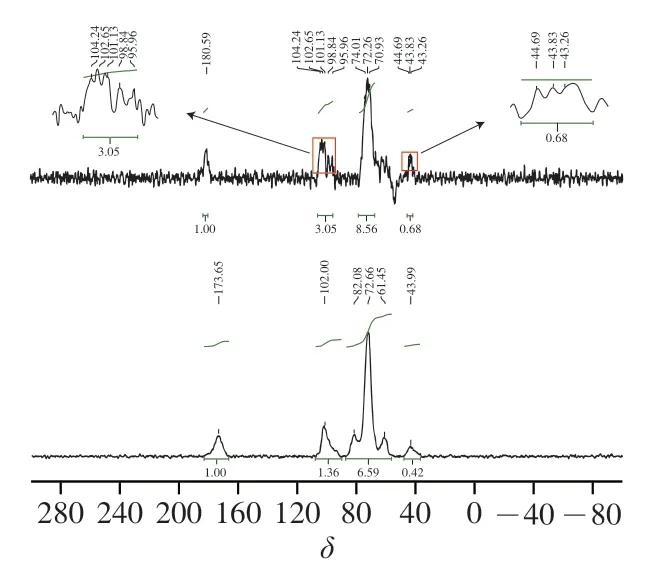
图8 MB-RS4·Se(IV)、MB-RS4 13C-NMR谱图Fig.8 13C-NMR spectra of MB-RS4·Se (IV) and MB-RS4
硒化改性后NMR氢谱中MB-RS4·Se(IV)的异头氢化学位移出现在5.01,硒化后化学位移大于5.00,说明硒化导致构型转变为构型。C谱中,180.59处为糖醛酸的典型信号峰。位于102.00处C1峰发生裂分出现104.24信号峰,可能是由于糖环在C1处由于Se的取代造成裂分并出现-异位形式的典型信号峰,硒化过程中1-4糖苷键断裂可能是造成糖环上C1羟基发生翻转造成构型改变的主要原因,位于82.09处C4的信号峰消失也验证了MB-RS4·Se(IV)中1,4-糖苷键发生断裂,1,4-糖苷键断裂后造成V型结晶区消失。NMR氢谱和碳谱中均表现出构型翻转与V型结晶区消失现象与红外谱图和X射线衍射图谱一致。位于MB-RS4·Se(IV)中C2/C3化学位移未发生较大改变和裂分,说明反应未发生在糖环的C2和C3。MB-RS4·Se(IV)中C6信号峰位移至70.93处,说明在C6位处发生了取代反应,直接验证了红外光谱中亚硒酸与C6上的羟基反应生成Se—O—C,Yuan Bo、Lee等研究发现,硒化多糖位于62.93和62.41的C6均发生了硒化取代,与本研究结果一致。
2.2 MB-RS4、MB-RS4·Se(IV)体外酶活抑制及动力学分析
2.2.1 对-葡萄糖苷酶和-淀粉酶抑制作用
-淀粉酶抑制剂和-葡萄糖苷酶抑制剂可以有效降低小肠内葡萄糖或果糖释放速率,从而延缓餐后血糖快速升高。图9为MB-RS4、MB-RS4·Se(IV)对-葡萄糖苷酶和-淀粉酶的抑制作用曲线,以阿卡波糖为阳性对照。MB-RS4对-葡萄糖苷酶抑制作用随质量浓度增加呈现负抑制率,不表现抑制作用。但对-淀粉酶具有显著的抑制活性,低于MB-RS4·Se(IV)和阳性对照阿卡波糖。其中MB-RS4和MB-RS4·Se(IV)的IC值分别为4.992 mg/mL和2.684 mg/mL。随着质量浓度升高,MBRS4·Se(IV)对-葡萄糖苷酶和-淀粉酶抑制作用升高后稳定,且呈现剂量依赖。硒化后的MB-RS4在同等浓度下对-葡萄糖苷酶表现显著抑制活性。以上数据表明,硒化改性使得对-葡萄糖苷酶无抑制作用的MB-RS4具有-葡萄糖苷酶抑制作用,同时MB-RS4·Se(IV)对-淀粉酶的抑制作用明显高于MB-RS4。MB-RS4·Se(IV)对-葡萄糖苷酶表现为抑制作用可能由于MB-RS4·Se(IV)中硒元素的引入。MB-RS4·Se(IV)对-淀粉酶抑制作用升高可能与MB-RS4·Se(IV)分子质量的降低有关,分子质量降低利于与酶的活性位点结合,导致抑制作用升高,这与MB-RS4·Se(IV)分子质量降低吻合。因此,需要通过抑制动力学分析研究其对-葡萄糖苷酶和-淀粉酶的抑制类型。
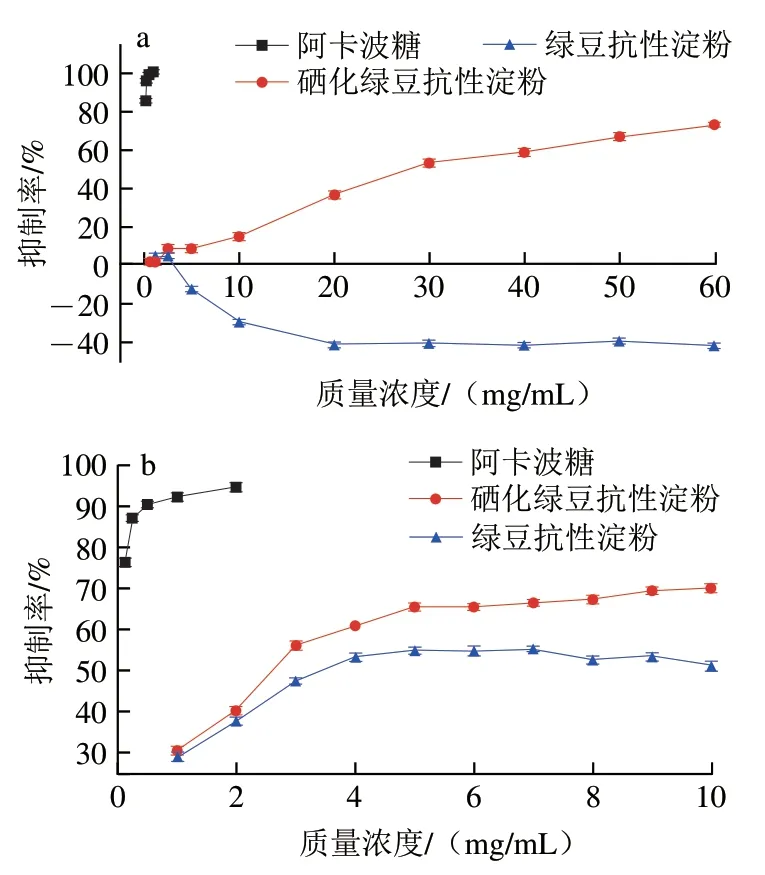
图9 MB-RS4、MB-RS4·Se(IV)对α-葡萄糖苷酶(a)和α-淀粉酶(b)的抑制作用Fig.9 Inhibitory effect of MB-RS4 and MB-RS4·Se (IV) on α-glucosidase (a) and α-amylase (b)
2.2.2 对-葡萄糖苷酶和-淀粉酶抑制动力学分析
根据Lineweaver-Burk曲线分析MB-RS4和MBRS4·Se(IV)对-葡萄糖苷酶和-淀粉酶的抑制类型。从图10a中MB-RS4·Se(IV)对-葡萄糖苷酶的酶促反应速率与不同质量浓度底物的双倒数图可知,随着MBRS4·Se(IV)质量浓度的增加,逐渐增大,逐渐减小,各质量浓度MB-RS4·Se(IV)的酶促反应速率拟合直线相交于第2象限。以上现象说明MB-RS4·Se(IV)对-葡萄糖苷酶为竞争抑制,MB-RS4·Se(IV)既可以通过与游离的-葡萄糖苷酶结合,也可通过与酶-底物复合物结合影响酶促反应的进行。图10b、c为MB-RS4和MB-RS4·Se(IV)对-淀粉酶的速率与底物质量浓度的双倒数图,随着MB-RS4和MBRS4·Se(IV)浓度的增加,逐渐减小,也随之减小,各浓度MB-RS4和MB-RS4·Se(IV)的酶促反应速率拟合直线相交于第3象限。说明MB-RS4和MB-RS4·Se(IV)对-淀粉酶为混合型反竞争性抑制,MB-RS4和MB-RS4·Se(IV)只能通过与酶-底物复合物的结合抑制酶促反应。
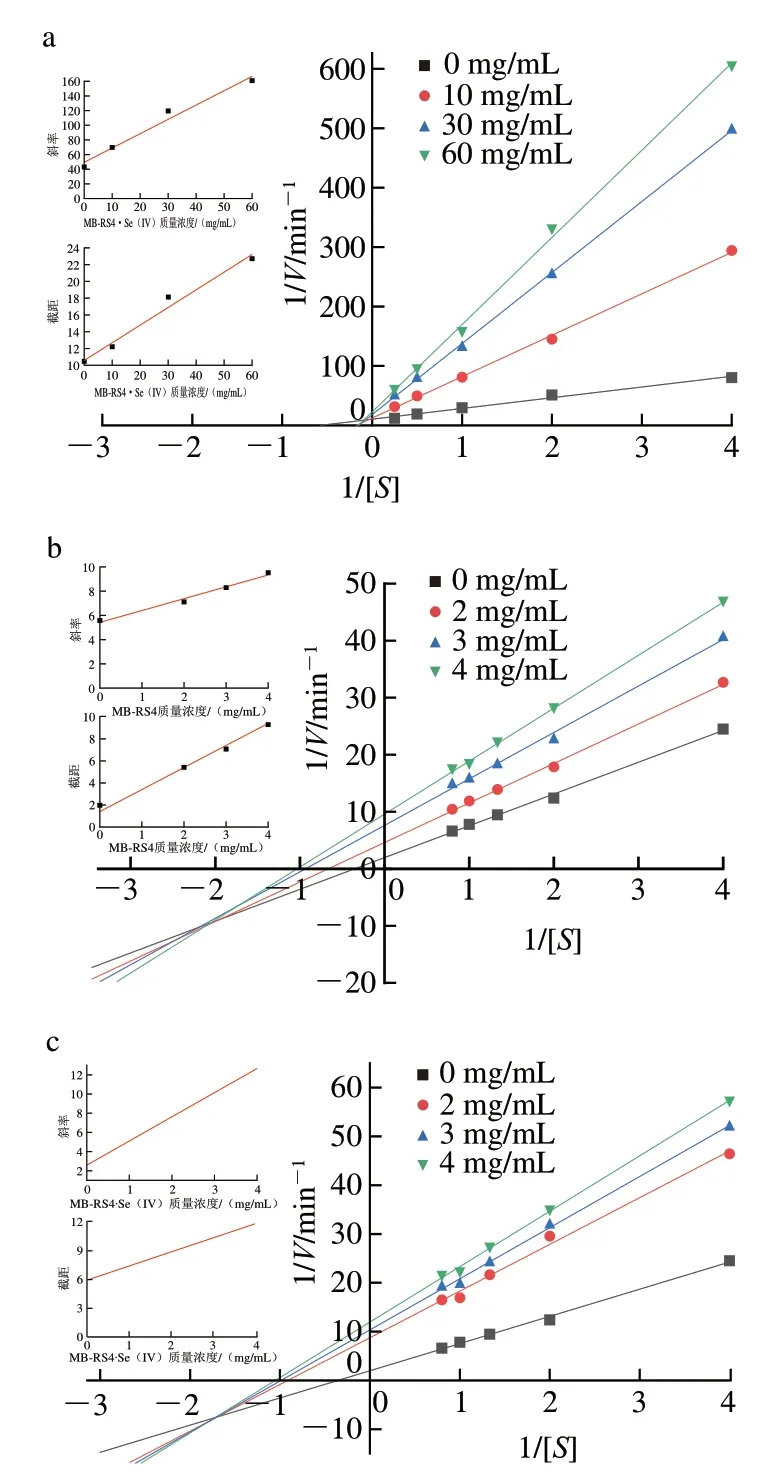
图10 MB-RS4·Se(IV)抑制α-葡萄糖苷酶(a)、MB-RS4抑制α-淀粉酶(b)、MB-RS4·Se(IV)抑制α-淀粉酶(c)的Lineweaver-Burk曲线图Fig.10 Lineweaver-Burk plots for α-glucosidase inhibition by MB-RS4·Se(IV) (a),and α-amylase inhibition by MB-RS4 (b) and MB-RS4·Se (IV) (c)
通过对比上图截距所反映的随样品质量浓度增加程度与抑制曲线发现,各质量浓度间截距的增长幅度与抑制曲线增长趋势基本吻合。同时由拟合的斜率与截距和各质量浓度MB-RS4、MB-RS4·Se(IV)的线性相关图发现其具有良好的线性关系,以上现象说明,MB-RS4和MB-RS4·Se(IV)结合在-葡萄糖苷酶、-淀粉酶和酶-底物复合物的单一抑制位点或单一类型的抑制位点上。
3 结论
对MB-RS4·Se(IV)结构进行表征及并研究其酶活抑制动力学,实验表明:MB-RS4·Se(IV)呈碎片化,表面粗糙,颗粒变小,出现Se=O、C—C(Se)、Se—O—C等官能团,紫外特征峰出现红移和增色效应,结晶结构完全消失,相对分子质量大幅度减小,NMR显示MBRS4·Se(IV)构型翻转并在C6上发生了取代反应。MB-RS4·Se(IV)对-葡萄糖苷酶表现抑制作用,对-淀粉酶抑制作用显著增加。通过酶促反应动力学实验分析得到MB-RS4·Se(IV)对-葡萄糖苷酶的抑制类型为竞争性抑制,对-淀粉酶的抑制类型为混合型反竞争性抑制。MB-RS4·Se(IV)可添加在降糖药物和功能性食品中,为减缓血糖升高协同微量元素强化提供新方向。
