活化及扩孔对正丁烷选择性氧化制顺丁烯二酸酐催化剂性能的影响
2022-10-26师慧敏张东顺
师慧敏,冯 晔,张东顺
(中国石化 北京化工研究院,北京 100013)
顺丁烯二酸酐(简称顺酐)的主要用途是生产玻璃钢行业的不饱和聚酯树脂,同时还可加氢生产1,4-丁二酸、γ-丁内酯、1,4-丁二醇及四氢呋喃。近年来,由于1,4-丁二酸和1,4-丁二醇作为生物可降解材料的单体广泛使用,顺酐产业再次引起人们的关注。目前,顺酐的工业原料已基本由苯转变为正丁烷,高性能钒磷氧(VPO)催化剂作为正丁烷法工艺的关键技术,需求愈加迫切[1]。对于固定床VPO催化剂来说,除催化剂本身活性中心价态、晶相组成及金属助剂种类[2-5]外,成型催化剂中活性中心与反应物的接触也直接影响催化剂的性能,特别是在实际生产中,促进自成型催化剂与高流速反应物分子接触[6-7]是提高催化剂使用效率的有效途径。同时,对于VPO催化剂,床层热点温度不应超过470 ℃,否则有可能在长周期运转下发生持续的不可逆催化剂相转变,从而影响催化剂的使用寿命[8]。因此,如何同时提高催化剂活性及选择性,并改善催化剂在床层中的传热性能是催化剂工业化过程中需要考虑的问题。
本工作采用有机溶剂还原法合成了VPO催化剂前体,在不同温度下活化后得到VPO催化剂,采用XRD、N2吸附-脱附、SEM、Raman光谱和XPS等方法对催化剂进行了表征;在VPO催化剂中加入扩孔剂,并用溶出的方法对催化剂成型体进行扩孔,研究了活化温度对催化剂晶相组成与催化性能的影响以及成型体中孔道形成对催化剂性能的影响。
1 实验部分
1.1 主要试剂
V2O5:分析纯,天津大茂化学试剂厂;草酸、磷酸:分析纯,西陇化工股份有限公司;异丁醇、丙酮、石墨粉:分析纯,国药集团化学试剂有限公司;1,1,1,-三羟甲基乙烷(TME):化学纯,Alfa Aeser(中国)化学有限公司。
1.2 催化剂制备
在双层玻璃反应釜中加入定量V2O5、草酸及异丁醇,搅拌使固体原料分散,向反应体系中逐步滴加定量纯磷酸。滴加完毕后,反应体系加热回流,得到浅蓝色浆液。将该浆液趁热抽滤、洗涤及干燥,得到的固体粉末与定量石墨粉混合后在压片机上压制为圆柱体。将该圆柱体置于管式反应炉中,向反应炉中通入体积比为1∶1的空气和水蒸气活化气氛,以4 ℃/min的升温速率分别升至400,415,425,440 ℃,并保持一定时间后,将活化气氛更换为体积比为1∶1氮气和水蒸气,继续在此温度和气氛条件下保持数小时,在氮气保护下降至室温。400,415,425,440 ℃活化的催化剂分别命名为VPO-Ⅰ,VPO-Ⅱ,VPO-Ⅲ,VPO-Ⅳ。将VPO-Ⅰ,VPO-Ⅱ,VPO-Ⅲ破碎成830 μm以下的颗粒后,向其中添加扩孔剂及石墨粉,混合均匀后冲压成异形圆柱成型体。将成型后的催化剂在定量丙酮中常温浸泡后滤出,真空干燥,得到的成型催化剂分别命名为VPO-Ⅴ,VPO-Ⅵ,VPO-Ⅶ。
1.3 催化剂表征
采用荷兰帕纳科公司的Empyrean型X射线衍射仪对催化剂晶相进行表征,步长为0.01°,2θ=5°~80°,CuKα射线(λ=0.15406 nm)。采用日本日立公司的Hitachi S-4800型冷场发射扫描电子显微镜对催化剂微观晶粒形貌及尺寸进行表征,加速电压为30 kV。采用美国Micromeritics仪器有限公司的ASAP-2020 型吸附仪对试样的比表面积、孔体积及孔结构参数进行测定,试样先在200 ℃抽真空处理4 h,至真空度达到0.67 Pa后开始测定。采用法国Jobin Yvon公司的HR-800型拉曼光谱仪对试样中V4+及V5+的结构特征光谱进行测定,室温,激光光源为532 nm,入射光功率为1.0 mW。采用英国VG公司的ESCALAB MK型光电子能谱仪进行XPS表征,以C 1s(284.8 eV)作为电子结合能校正标准,分析室真空度小于2×10-6Pa,MgKα射线,能量为1253.6 eV。采用瑞士万通公司的916 Ti-Touch型自动滴定仪对催化剂钒中心的价态进行测试,高锰酸钾和硫酸亚铁铵为标准溶液。
1.4 催化剂性能评价
不同活化温度及扩孔前后的成型催化剂的性能均在120 mL单管反应器中进行评价,反应管内径为21 mm,内置外径为4 mm的温度套管。催化剂装填高度约为50 cm。空气与正丁烷分别计量后进入反应管中,在活化后的VPO催化剂作用下选择性氧化生成顺酐,尾气出口处加装支路,测定其中的CO和CO2含量,通过排水法收集一定体积尾气中存在的顺酐。采用美国安捷伦公司的7890B型气相色谱仪检测气相组成,采用FID测定原料气及尾气中的正丁烷含量,计算正丁烷转化率;采用TCD测定尾气中CO与CO2的含量。顺酐含量用标准碱液滴定确定。正丁烷、CO及CO2含量分别由正丁烷、CO及CO2标气标定所得。
2 结果与讨论
2.1 活化温度对催化剂性能的影响
在对成型催化剂的孔结构进行调变前,确定成型的催化剂粒子具有稳定的晶相组成及一致的V4+/V5+。首先,将V2O5与磷酸(P与V原子比为1.15)混合于异丁醇中,回流条件下发生还原反应,体系逐渐由初始的棕色转变为墨绿色,最终形成浅蓝色的混合体系。将此混合体系分离、干燥后得到催化剂前体磷酸氢氧钒半水化合物(VHP),VHP在不同温度下活化后的XRD谱图见图1。由图1可知,VHP主要的晶面衍射峰出现在2θ=15.6°,19.7°,24.3°,27.1°,28.8°,30.4°,37.5°,49.2°处,分别对应于VHP的(001),(101),(021),(121),(201),(220),(040),(331)晶面[9],(220)晶面衍射峰强度较强,其他衍射峰强度较弱,表明此时生成的VHP(220)晶面为优势晶面。一般来说,V2O5与磷酸反应后会较快形成VOPO4·2H2O。催化剂制备过程中采用异丁醇作为还原剂,还原能力弱且空间位阻较小,在与VOPO4·2H2O作用过程中,易接近钒氧中心,从而有利于平行z轴(220)晶面的生成[10]。同时,在V2O5与磷酸反应后的液体介质中明确检测出异丁醛,说明在V2O5还原生成VHP的过程中,异丁醇被氧化为相应的醛。将干燥后的亮蓝色前体VHP在250 ℃焙烧后,试样的晶相并未发生明显变化,主要晶相仍为VHP。

图1 催化剂前体VHP在不同温度活化后的XRD谱图Fig.1 XRD patterns of the VOHPO4·0.5H2O(VHP) precursor activated at different temperatures.
将焙烧后的前体与定量石墨混合,压制成坯块进行高温活化。O’Mahony等[11]研究发现,在活化过程中,半水化合物在300 ℃开始分解为无定形固体,350 ℃开始出现新相,400 ℃新相完全形成,VHP逐渐完成了向活性相的拓扑转变。活化温度为400 ℃时,炉中催化剂表面实际温度较加热温度高20~30 ℃,最高温度为431 ℃。由图1还可知,VPO-Ⅰ晶面衍射峰出现在2θ=14.1°,15.6°,18.5 °,23.0 °,28.5 °,29.9 °,33.7 °,36.8 °,43.4°,46.9°,49.4°处,分别对应(VO)2P2O7(VPP,JCPDS No.34-1381)的(102),(012),(200),(020),(204),(221),(106),(125),(008),(040),(512)晶面,其中,(020)和(204)晶面分别由原VHP中的(001)和(101)晶面演变而来。另外,VPO-Ⅰ催化剂除在活化后大量形成V为+4价的VPP外,V为+5价的VOPO4化合物含量较少,未形成明显晶相。控制活化温度至415 ℃,VPO-Ⅱ表面最高温度为442 ℃,除VPP外,逐渐形成含有VOPO4的混合晶相,其中,2θ=19.5°,22.1°,24.2°处的衍射峰分别对应δ-VOPO4(JCPDS No. 47-0951)的(002),(111),(012)晶面,说明随活化温度的升高,VOPO4含量明显增加。δ-VOPO4的出现可能是由于在接近450 ℃时,VPP与δ-VOPO4的相互转化[12],而且当温度达到450 ℃时,VHP在含氧气氛中可直接生成δ-VOPO4。控制活化温度至425 ℃,VPO-Ⅲ表面最高温度可达到451 ℃,此时除观察到δ-VOPO4的特征衍射峰外,2θ=24.8°,29.0°,29.7°处衍射峰的出现说明活化后的催化剂中同时存在另外一种V为+5的价VPO晶相,分别对应αII-VOPO4(JCPDS No.34-1247)的(101),(111),(200)晶面。继续提高活化温度至440 ℃,VPO-Ⅳ表面最高温度可达480 ℃,2θ=22.0°处的衍射峰强度明显增强,归属于δ-VOPO4的(111)晶面,说明此时δ-VOPO4成为最主要的晶相。随着活化温度的升高,VOPO4晶相明显增多。一般来说,V5+在反应中的存在是必要的,研究者已普遍认同需要V4+与V5+共存,才能活化正丁烷的C—H键并进一步选择性生成顺酐[13-15],但这种有效的V5+来自于与(VO)2P2O7活性相相互作用的V5+无定形微畴或微晶[14],或来自于(VO)2P2O7活性相表面高度离散分布的V5+中心[15],而不是来自于独立成相的VOPO4晶相。当催化剂表面温度达到450 ℃以上时,V5+晶相成为明显可见的晶相,甚至在更高温度时成为主要晶相。占据主要晶相组成的V5+晶相的存在会改变VPO催化剂的堆垛方式,V5+晶相与含氧化合物顺酐的作用加强,从而造成活性位上的顺酐不易脱离,过度氧化成COx[16-17]。因此,在活化过程中控制催化剂表面温度在450 ℃以下,可以抑制V5+中心过多地独立成相。
VHP活化前后的SEM照片见图2。由图2可知,VHP发生拓扑转变时,晶粒的形貌和表观颜色发生明显变化。VHP呈不规则片状结构,部分片层发生一定弯曲,片层尺寸不均一,这也是导致晶面衍射峰强度较低的原因(见图2a)。在250 ℃焙烧后,粉末变为灰黑色,前体虽然发生比较明显的碎裂,片层尺寸减小,但此时片状结构仍得以保持(见图2b)。随着温度的升高,前体破碎成更细小的片层从而进行晶相转变,VHP先转变为部分氧化的无定形相后继续活化为高度结晶的VPP。经此过程后,催化剂表观颜色变为深绿色,催化剂晶粒在原聚集体的形貌上发生明显的破碎细化,晶粒粒径均小于50 nm,这是活性VPP的典型形貌(见图2c)。催化剂晶粒细化有利于提高催化正丁烷氧化的活性。同时当活化后的催化剂中含有大量的VOPO4晶相时(见图2d),催化剂表观颜色为黄绿色,晶粒尺寸均一性降低,且颗粒粒径增大至约150 nm,这会直接影响催化剂活性。

图2 VHP活化前后的SEM照片Fig.2 SEM images of VHP before and after activation. a VHP;b VHP calcinated at 250 ℃;c VPO-Ⅲ;d VPO-Ⅰ
对表面温度在450 ℃以下或接近450 ℃的催化剂试样的表面物种组成进行Raman光谱分析,结果见图3。
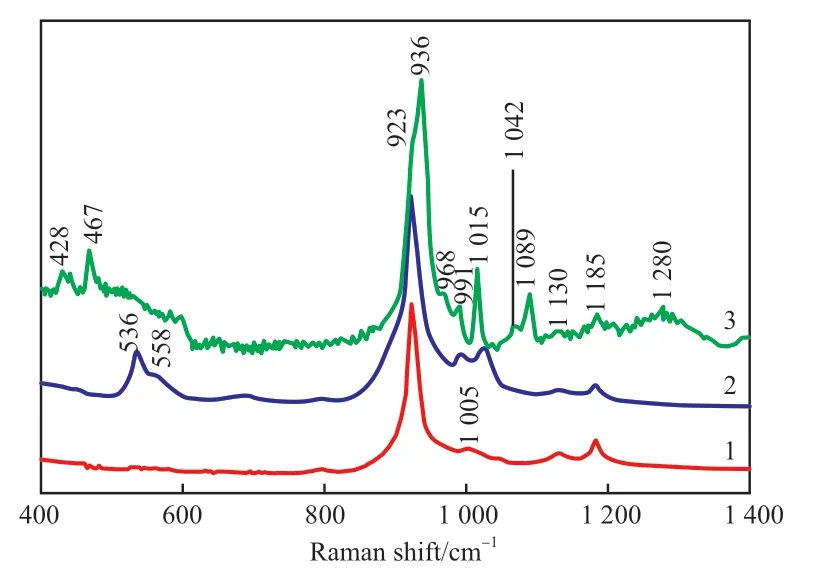
图3 不同活化温度的VPO催化剂的Raman谱图 Fig.3 Raman spectra of VPO catalysts activated at different temperatures. 1 VPO-Ⅰ;2 VPO-Ⅱ ;3 VPO-Ⅲ
由图3可知,VPO-Ⅰ中主要存在的化合物为VPP,923,1130,1185 cm-1处的吸收峰为VPP的特征峰,其中,923 cm-1处的吸收峰归属于焦磷酸根中P—O—P的伸缩振动,1185 cm-1处的吸收峰归属于V—O—P的伸缩振动,1015 cm-1处肩峰的存在证实催化剂表面有极少量的δ-VOPO4微晶,它归属于δ-VOPO4上V=O键的伸缩振动;同时,1042 cm-1处较弱的吸收谱带属于V5+的V=O键的特征峰,它的出现说明VPP母体上存在比较孤立的V5+中心[18]。VPO-Ⅱ中除(VO)2P2O7晶相外,δ-VOPO4的分立越来越明显,即升高温度进一步促进了(VO)2P2O7向δ-VOPO4的转变。VPO-Ⅲ中V5+物种的含量进一步增加,除δ-VOPO4外,968,1089,428,467 cm-1处出现归属于αII-VOPO4的特征吸收峰,说明随着温度的升高,催化剂中部分(VO)2P2O7继续进行相转变,进一步形成δ-VOPO4和αII-VOPO4。活化过程中控制催化剂表面实际温度低于450 ℃,可以起到抑制表面V5+物种大量生成的作用。
对VPO-Ⅰ~VPO-Ⅲ的表面钒中心电子结合能及价态进行分析,V 2p3/2拟合图见图4。由图4可知,活化后催化剂的V 2p3/2峰均发生分裂,形成明显的V4+和V5+中心。VPO-Ⅰ中V 2p3/2的电子结合能为517.4,518.3 eV,表面n(V4+)∶n(V5+)=62.5∶37.5,氧化还原滴定法测定钒价态为4.13,尽管XRD和Raman谱图均显示活化后的催化剂中主要组分为VPP,但实际上催化剂中仍含有部分V5+物种,此物种以无定形或高度离散的孤立V5+中心存在。VPO-Ⅱ中V 2p3/2的电子结合能为517.2,518.7 eV,n(V4+)∶n(V5+)=60.3∶39.7,钒价态为4.23,钒中心整体价态升高。与较低活化温度的催化剂相比,两者之间相近的n(V4+)∶n(V5+)说明催化剂表面的V5+物种含量相近,XRD和Raman谱图证实,VPO-Ⅱ中存在明显的δ-VOPO4,V5+物种更多以聚集态的微晶或微畴的形式存在。VPO-Ⅲ中,V 2p3/2的电子结合能为516.7,517.7 eV,n(V4+)∶n(V5+)=46.1∶53.9,钒价态为4.37。与其他两个催化剂相比,VPO-Ⅲ上钒中心电子结合能下降可能归因于较为明显的δ-VOPO4和αII-VOPO4独立成相现象,催化剂表面上V4+与V5+中心之间的相互作用减小,不利于催化剂活性及选择性的提高。

图4 不同活化温度的VPO催化剂的XPS V 2p3/2拟合图Fig.4 XPS fitting results of V 2p3/2 for VPO catalysts activated at different temperatures.
不同活化温度的VPO 催化剂的评价结果见表1。由表1可知,在正丁烷含量1.6%~1.8%(φ)、气态空速1500 h-1、盐浴温度400 ℃的条件下,不同活化温度的催化剂表现出明显不同的初始反应性能。VPO-Ⅰ表现出中等的催化活性,正丁烷转化率为79%~80%,顺酐选择性为70%~71%;VPO-Ⅱ表现出略高的反应活性,正丁烷转化率约为80%,顺酐选择性略有降低,为66%~68%;VPO-Ⅲ的活性最高,正丁烷转化率增至83%~85%,但顺酐选择性明显下降,仅为62%左右。比较3种催化剂的晶相组成,当催化剂中含有较多的V5+独立晶相或微晶时,正丁烷转化率较高而顺酐选择性略低,而催化剂中含有较大量的V4+,V5+中心为高度离散的孤立V5+时,催化剂表现出较高的顺酐选择性。

表1 不同活化温度的VPO催化剂的评价结果Table 1 Catalytic results of VPO catalysts activated at different temperatures
2.2 扩孔对催化剂性能的影响
分别对以上具有不同晶相组成及性能的催化剂进行扩孔。在催化剂颗粒中添加TME,通过缓慢溶解的方式将TME逐渐从成型体中溶出,制造催化剂内孔道。扩孔前后催化剂的孔结构参数及机械强度见表2。由表2可知,扩孔剂溶出后,催化剂的比表面积并不是全部呈增大的趋势。VPO-Ⅰ扩孔后形成VPO-Ⅴ,比表面积增大15.5%,孔体积增大20.2%,最可几孔径由13.7 nm增至20.2 nm。随着孔体积的增大,VPO-Ⅴ中2~50 nm的介孔比例略有下降,从83.2%降至79.7%,大孔比例由15.3%增至19.8%,同时介孔中2~20 nm的孔道比例由30.7%增至46.1%,说明该催化剂孔体积和比表面积的变化主要来自于原来部分微孔或无孔区域被扩大成介孔。VPO-Ⅱ扩孔后形成VPO-Ⅵ,比表面积降至原来的92%,但孔体积却提升73.8%,同时最可几孔径由6.9 nm增至16.4 nm,大孔比例明显增加,由8.8%增至20.8%,而介孔中20~50 nm的孔的比例由61.4%降至30.5%,比表面积的下降主要来自于更多的介孔被扩大成大孔。VPO-Ⅲ扩孔后形成VPO-Ⅶ,比表面积较之前增大84%,孔体积增大157%,最可几孔径由6.6 nm增至12.9 nm,介孔中2~20 nm的孔的比例由37.8%增至90.6%,比表面积的显著增大主要来自于这部分介孔的增多。这说明添加扩孔剂可以明显改变催化剂的孔结构:一方面,由于扩孔剂的使用明显增大了催化剂的孔体积和最可几孔径,最可几孔径从扩孔前的6.6~13.7 nm增至扩孔后的12.9~20.2 nm,小于2 nm左右的微孔含量明显减少,孔径及孔体积的增大有利于反应物分子在催化剂表面及孔道中的扩散;另一方面,添加扩孔剂可以改变催化剂的比表面积,扩孔后由于催化剂中各种孔道所占比例发生变化,催化剂的比表面积发生改变,总体保持较高的比表面积,这有利于反应分子与更多催化剂活性位的接触,从而进一步改善催化剂的活性。由表2还可知,将扩孔剂从催化剂本体中溶出后,通过氧化还原滴定,发现钒中心的氧化态基本保持不变,说明通过溶解扩孔剂扩孔的物理扩孔方式不会影响催化剂活性中心的价态。同时还发现,由于大量的扩孔剂从催化剂成型体中溶出,VPO-Ⅴ,VPO-Ⅵ,VPO-Ⅶ的侧向抗冲压强度分别下降了15.6%,18.2%,31.3%。虽然最初冲压时控制成型催化剂的侧向抗冲压强度在30~35 N,但孔道的形成一定程度上降低了成型催化剂的机械强度,从而可能会影响固定床床层催化剂的长期结构稳定性。

表2 VPO催化剂的物性Table 2 Physical parameter of VPO catalysts
扩孔前后VPO催化剂的XRD谱图见图5。由图5可知,催化剂扩孔后无新的特征衍射峰出现,说明扩孔后无新晶相生成,催化剂的主要组成没有变化。石墨的特征衍射峰在扩孔后明显变小,这是由于石墨具有极强的流动性,在溶出扩孔剂的同时也有部分石墨逃逸。

图5 扩孔前后VPO催化剂的XRD谱图Fig.5 XRD patterns of the VPO catalysts before and after pore expanding.
扩孔前后催化剂的Raman谱图见图6。由图6可知,VPO-Ⅰ扩孔后得到的VPO-Ⅴ的主要组成仍为VPP,924,1132,1186 cm-1处的吸收峰归属于VPP的特征峰;VPO-Ⅱ扩孔后,VPO-Ⅵ在1016,1070 cm-1处的肩峰更加明显,证实催化剂表面少量的δ-VOPO4微晶的分立越来越明显,1088,428,467 cm-1处出现αII-VOPO4的特征峰,说明扩孔后有较多的αII-VOPO4暴露;VPO-Ⅲ扩孔后,VPO-Ⅶ中V5+物种的特征峰愈加明显。同时还发现,扩孔后的催化剂VPO-Ⅵ和VPO-Ⅶ在924 cm-1处的特征峰出现分裂,935 cm-1处较为明显的肩峰归属于δ-VOPO4物种中的对称伸缩振动[19],这可能是由于扩孔后暴露了更多的δ-VOPO4物种。结合XPS、XRD和Raman光谱分析结果,虽然扩孔后催化剂的晶相组成及表观价态未发生变化,但表面物种分布发生了改变。较多的V5+中心出现在催化剂表面,可能对催化剂的性能有一定影响。
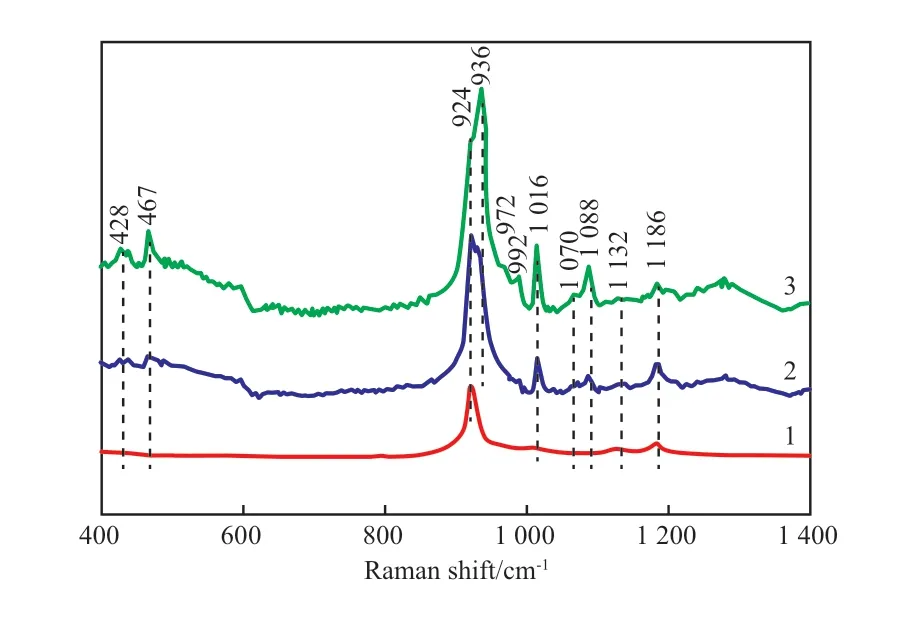
图6 扩孔后VPO催化剂的Raman谱图Fig.6 Raman spectra of the VPO catalysts after pore expanding. 1 VPO-Ⅴ;2 VPO-Ⅵ;3 VPO-Ⅶ
扩孔后的VPO 催化剂的评价结果见表3。由表3可知,在气态空速1500 h-1、正丁烷含量1.5%~1.8%(φ)、盐浴温度400 ℃的条件下,VPO-Ⅴ在初始反应阶段表现出较高的正丁烷转化率(大于80.0%)及较高的顺酐选择性(大于等于70.0%)。与未扩孔VPO-Ⅰ相比(见表1),正丁烷转化率明显提高,同时顺酐选择性得以保持(70.0%~73.0%)。从催化剂孔道结构看,该催化剂在扩孔前即具有较为适宜的最可几孔径(13.7 nm),扩孔后大孔含量的增多和孔体积的增大有利于进一步发挥孔道内活性中心的作用,因此,正丁烷转化率提高。同时,由于催化剂自身晶相组成中较为适宜的V4+,V5+种类及比例,催化剂整体性能提高。在提高顺酐收率的同时,催化剂床层的热点温度降至436~440 ℃。与未扩孔VPO-Ⅱ相比,VPO-Ⅵ的正丁烷转化率略有上升,但生成顺酐的选择性基本保持不变(66%~68%)。究其原因,可能是由于扩孔后孔体积的增大主要来自于更多的介孔被扩成大孔,因此,反应分子较易与孔道中的活性中心接触,但同时由于催化剂孔道中暴露了更多的V5+非活性晶相,正丁烷转化率提高有限。VPO-Ⅶ在扩孔前即表现出高活性、低选择性的特点,由于催化剂存在多种V5+晶相,特别是非选择性晶相αII-VOPO4的存在导致较多副反应发生,造成COx含量较高;虽然VPO-Ⅶ中2~20 nm介孔孔道明显增多,但大孔孔道明显减少;反应物扩散到较小的介孔孔道后,受催化剂自身晶相组成影响,较易发生副反应,导致顺酐选择性下降;而进一步提高正丁烷含量时,虽然催化剂有较高的比表面积,但反应物优先与较少量的大孔孔道中的活性中心接触,未利用到介孔孔道中更多的活性中心,导致正丁烷转化率下降;相比扩孔前催化剂的性能(顺酐收率52.0%~53.0%(x)),扩孔后催化剂的顺酐收率基本保持不变,为47.0%~54.0%(x)。
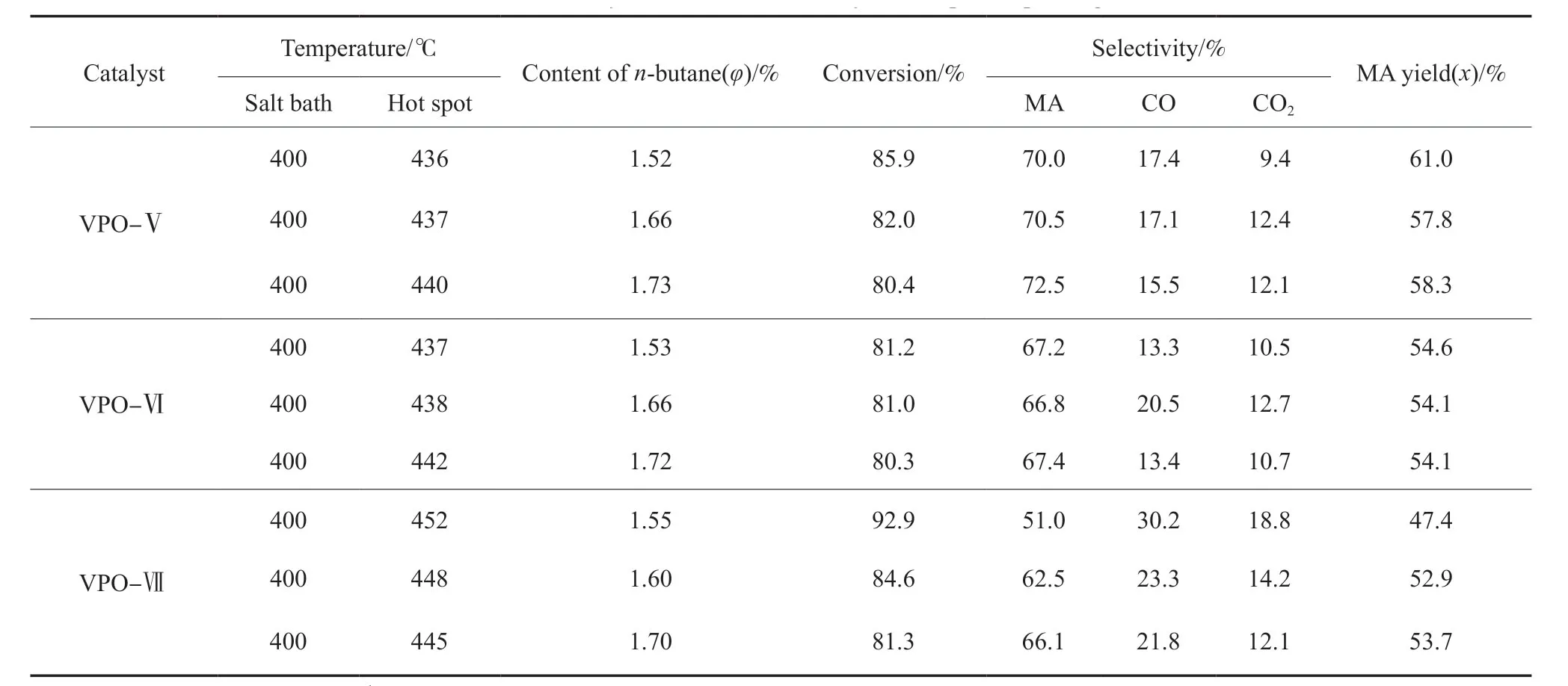
表3 扩孔后VPO催化剂的评价结果Table 3 Catalytic results of VPO catalysts after pore expanding
相较于未扩孔的催化剂,在相同反应条件下,采用扩孔后的催化剂催化反应时,热点温度均下降2~10 ℃,说明扩孔后催化剂的孔结构有明显改进,有效抑制了过氧化副反应的发生。同时径向催化剂床层热点与盐浴的温度差进一步降低,也说明对催化剂进行扩孔有利于催化剂床层的传热。从以上分析结果可看出,反应中大孔孔道比例的增加有利于提高催化剂活性中心的利用率,对催化剂晶相组成进行控制后,对催化剂成型体的孔结构进行进一步调控,扩大催化剂的孔体积及大孔孔道比例,可以有效提高顺酐收率。
3 结论
1)通过调控活化温度可以控制VPO催化剂前体在活化过程中的晶相组成,得到有利于正丁烷选择性氧化制顺酐反应的V4+及V5+中心分布。
2)通过对VPO催化剂进行扩孔,使催化剂的孔体积、最可几孔径、介孔和大孔比例都发生了明显变化,可以较好地调节反应物及生成物分子在催化剂表面的扩散,从而提高正丁烷转化率。
3)适当添加扩孔剂,使VPO催化剂的大孔比例增加,有利于提高催化剂活性中心的利用率,特别是在适宜的催化剂晶相组成下,可明显提高顺酐收率。
