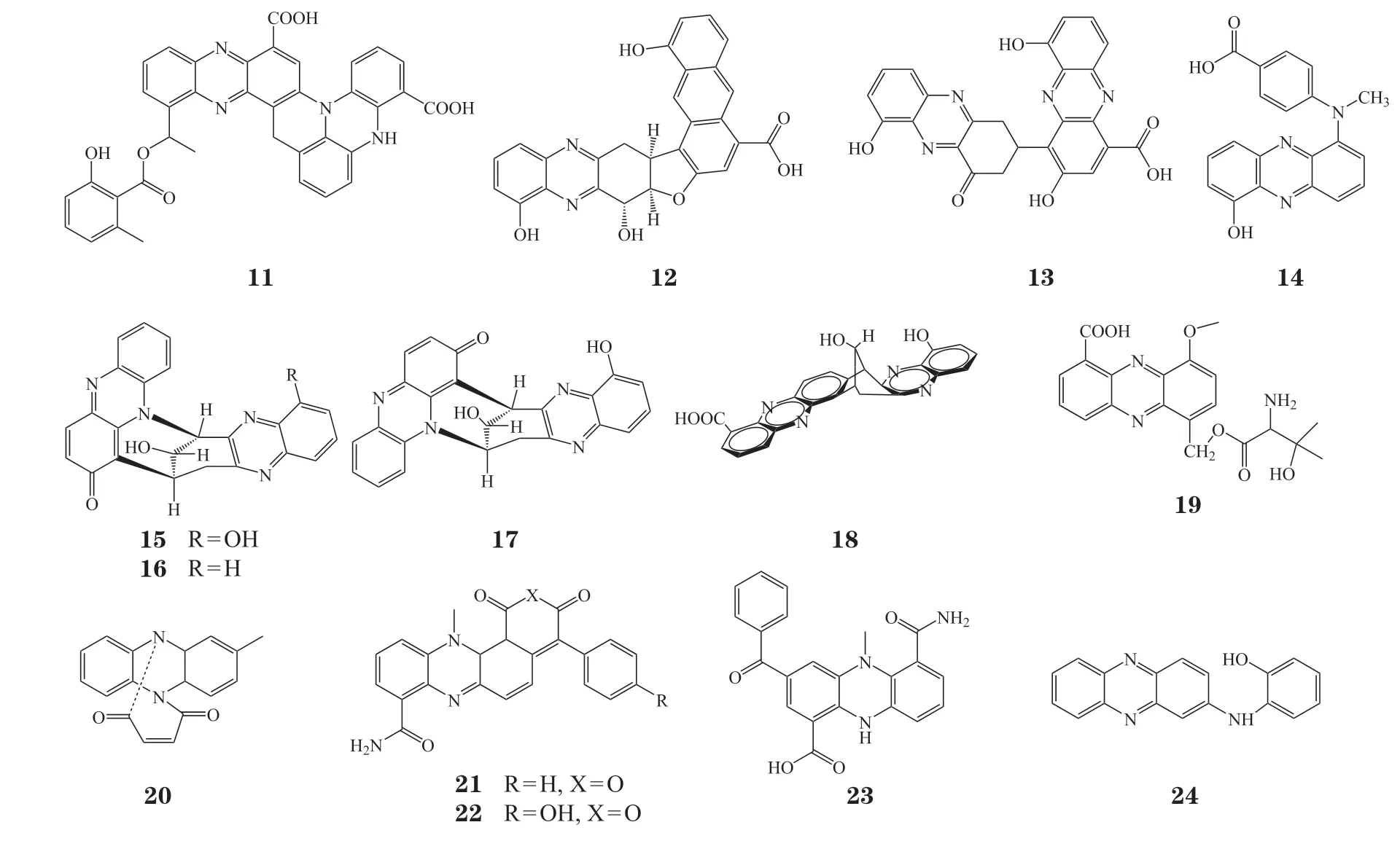
吩嗪及其类似物抗肿瘤活性研究进展
2022-10-14刘静徐幼桥张春花夏卓璐王志祥黄德春江峰
刘静,徐幼桥,张春花,夏卓璐,王志祥,黄德春,江峰*
(1.中国药科大学工学院制药与环境工程系,江苏 南京 211198;2.南京大学医学院附属鼓楼医院医院感染管理办公室,江苏 南京 210008)

天然来源的吩嗪及其衍生物是一类源于土壤或海洋环境中的链霉菌、假单胞菌和海洋微生物的次生代谢产物,具有很强的氧化还原能力,表现出抗菌、抗病毒、抗疟和抗肿瘤等生物活性。目前已报道有100余种天然吩嗪产物和6 000余种吩嗪合成类似物[1-3]。自20世纪50年代人们发现吩嗪具有抗肿瘤活性以来,国内外已有不少关于抗肿瘤吩嗪及其类似物的论文发表,但迄今国内缺乏对该类化合物在抗肿瘤药物研发领域的进展总结。本文将对天然来源的吩嗪及其衍生物以及合成类似物的研究现状进行综述,重点关注该类化合物的结构、抗肿瘤活性和作用机制的研究进展,以期为该类化合物在药物研发中的应用提供参考。
1 天然来源的吩嗪及其衍生物
1.1 从假单胞菌分离得到的天然吩嗪衍生物
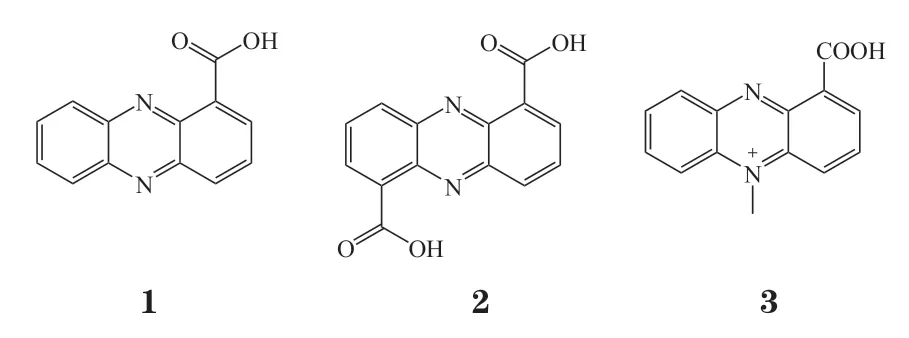
吩嗪-1-羧酸(phenazine-1-carboxylic acid,PCA,1)和吩嗪-1,6-二羧酸(phenazine-1,6-dicarboxylicacid,PDA,2)是最早被发现具有抗菌活性的天然吩嗪衍生物[4-5]。Dasgupta等[6]从HRW.1-S3铜绿假单胞菌中分离出PDA,研究表明PDA以剂量依赖性方式对HT29、HeLa和MCF7肿瘤细胞表现出细胞毒性,而PCA 对HT29细胞也具有一定抗增殖活性,二者的IC50均为25 μmol · L-1。
Patil等[7]从海洋铜绿假单胞菌GS-33中分离得到微生物色素PCA,研究结果表明其对人皮肤黑色素瘤细胞株SK-MEL-2具有抑制增殖作用,细胞抑制活性(GI50)小于44.63 μmol · L-1。
Kennedy等[8]从根际土壤细菌PUW5中分离出吩嗪类化合物MPCAB(3),其对肺癌A549细胞和乳腺癌MDA-MB-231细胞具有优良的选择性增殖抑制作用,IC50分别为488.7和458.6 nmol · L-1,而对正常细胞PBMC无明显毒性。研究发现化合物3通过激活含半胱氨酸的天冬氨酸蛋白水解酶 3(cysteinyl aspartate specific proteinase,caspase-3)和下调B细胞淋巴瘤2(B-cell lymphoma 2,Bcl-2)蛋白从而诱导癌细胞凋亡。
1.2 从链霉菌分离得到的天然吩嗪衍生物
Tamaoki等[9]从链霉菌DO-59中分离得到PCA酰胺类似物4和5,研究发现它们对小鼠肉瘤180细胞具有显著的增殖抑制活性。

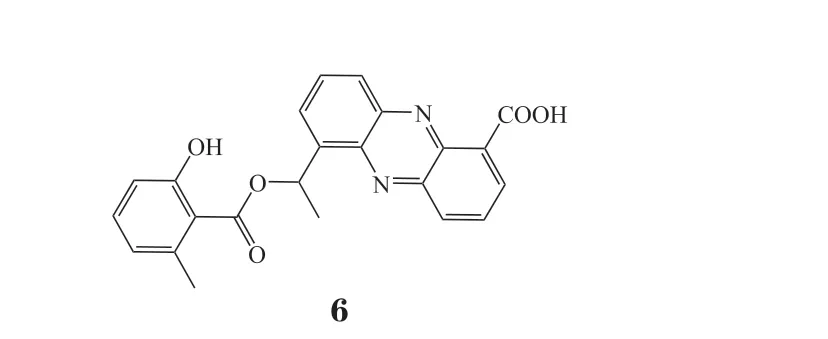
Kitahara等[10]从加那利链霉菌MG314-hF8中分离得到沙霉素(saphenamycin,6),其对小鼠白血病L5178Y细胞和L1210细胞具有增殖抑制活性,IC50分别为0.37和(1.48 ~ 6.19) μmol · L-1,对CCRF/CEMT白血病细胞的IC50为1.48 μmol · L-1[11]。
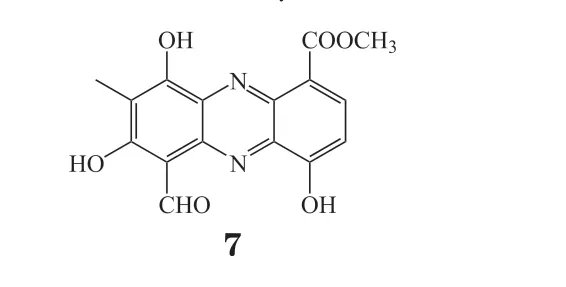
革兰氏阳性放线菌链霉菌具有产出多取代吩嗪的能力[1],从中分离出的六取代吩嗪PD 116,152(7)具有清除自由基和抗肿瘤活性[12],对HTC-8结肠腺癌细胞和L1210淋巴白血病细胞具有显著的细胞毒性,IC50分别为0.71和0.52 μmol · L-1[13]。
N-异戊二烯基化吩嗪霉素(8)是从链霉菌WK-2057.13中分离得到的首个结构中含有倍半萜结构的吩嗪酮生物碱。该化合物具有抗肿瘤活性,对阿霉素耐药的P388白血病细胞的IC50为7.46 μmol · L-1,对正常小鼠P388白血病细胞的IC50为62.15 μmol · L-1[14-15]。

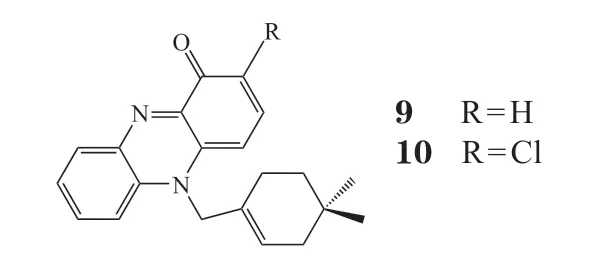
N-取代单萜类似物WS-9659A(9)和WS-9659B(10)从链霉菌中被分离得到,研究表明该类化合物对小鼠P388白血病细胞和L1210白血病细胞具有细胞毒性,IC50均为0.27 μmol · L-1。二者对大鼠、狗和人的前列腺细胞睾酮5α-还原酶活性具有抑制作用,其抑制作用呈剂量依赖性[16-17]。
Bahnmuller等[18]分离出吩嗪二聚体衍生物Esmeraldine B(11),其对CCRF/CEM人白血病细胞具有增殖抑制作用,IC50为0.63 μmol · L-1[11]。
肿瘤坏死因子相关凋亡诱导配体(TNF-related apoptosis-inducing ligand,TRAIL)具有选择性杀伤肿瘤细胞的作用[19-20]。Abdelfattah等[21]从链霉菌IFM 1204菌株中分离出3种新的吩嗪类似 物izumiphenazines A、izumiphenazines B和izumiphenazines C(12~14),其对TRAIL耐药的人胃腺癌细胞具有增殖抑制作用,IC50分别为40、30和20 μmol · L-1。化合物13和14与TRAIL联用对TRAIL耐药的AGS细胞具有协同增敏作用[21]。
Ding等[22]从土壤样品中分离出链霉菌菌株YIM DT26,研究发现其产生的次级代谢产物phenazinolins A、phenazinolins B和phenazinolinsC(15~17)对P388、GLC、H460和XWLC肿瘤细胞具有增殖抑制活性,IC50为14 ~ 40 μmol · L-1。
Li等[23]从链霉菌亚种中分离到一种新的吩嗪二聚体(18),该化合物对人肿瘤细胞株BGC823具有细胞毒性,IC50为14.9 μmol · L-1。
1.3 从其他菌属分离得到的天然吩嗪衍生物
海洋微生物被广泛认为是次生代谢物的重要来源[24]。1997年,Imamura等[25]从一种新的海洋细菌变异远洋杆菌属分离得到新的吩嗪类化合物,其中pelagiomicin A(19)对HeLa细胞具有细胞毒性,IC50为0.1 μmol · L-1。Singh等[26]研 究 发 现 化 合 物pelagiomicin A通过抑制DNA、RNA和蛋白质合成,对A2780S卵巢癌细胞和SW620结肠癌细胞具有细胞毒性,IC50分别为0.45和1.25 μmol · L-1。
Li等[27]从太平洋深海沉积物中采集的芽孢杆菌培养液中分离出一种含有[4,2,2]环结构的吩嗪天然产物20。研究结果表明,化合物20对P388细胞具有细胞毒性,在浓度为50 μmol · L-1时对P388细胞增殖抑制率达78.3%,而对K562细胞无毒性。
2010年,Abdel-Mageed等[28]从马里亚纳海沟沉积物中分离出放线菌菌株MT1.1和 MT1.2,并从其代谢物中分离得到一系列吩嗪天然产物,其中化合物21和22对白血病细胞株K562具有细胞毒性,IC50分别为9和7 μmol · L-1。化合物23呈现出较高的自由基清除活性,其IC50为8.4 μmol · L-1。
Gao等[29]从北冰洋深海采集的沉积样品中分离出放线菌菌株BM-17,并从其次生代谢物中分离得到吩嗪天然产物NHP(24),该化合物对HepG2、A549、HCT-116和COC1细胞具有一定的细胞毒性,IC50分别为40.33、38.53、27.82和28.11 μmol · L-1。
2 合成吩嗪类似物
基于吩嗪类化合物广泛的生物学特性和医药用途,人们对该类化合物的合成、结构修饰和结构-活性关系进行了大量研究。
2.1 吩嗪-1-甲酰胺合成类似物
吩嗪-1-甲酰胺(25)作为一种农药被人们所熟知,近年来研究者们开展了其结构类似物的药理活性研究[30]。Atwell等[31]发现了一种高效的拓扑异构酶Ⅰ/Ⅱ双重抑制剂DACA(26),在Ⅱ期临床试验中,由于其代谢快,在人体反应率低,于2001年终止研发[1,32-33]。该课题组将吩嗪-1-羧酸和胺偶联得到吩嗪-1-甲酰胺类似物27~31[34](见图1)。当化合物27中的n= 2 时得到化合物28,其对L1210、P388和Lewis细胞的增殖抑制活性较好。随后,他们以化合物28为先导化合物,设计合成了一系列吩嗪-1-甲酰胺类化合物。构效关系表明,9位上被Cl、CH3和OCH3取代的化合物29、30和31的细胞毒活性明显高于化合物28,其中化合物30对P388细胞的增殖抑制活性最好,IC50为18 nmol · L-1[33-34](见图1)。
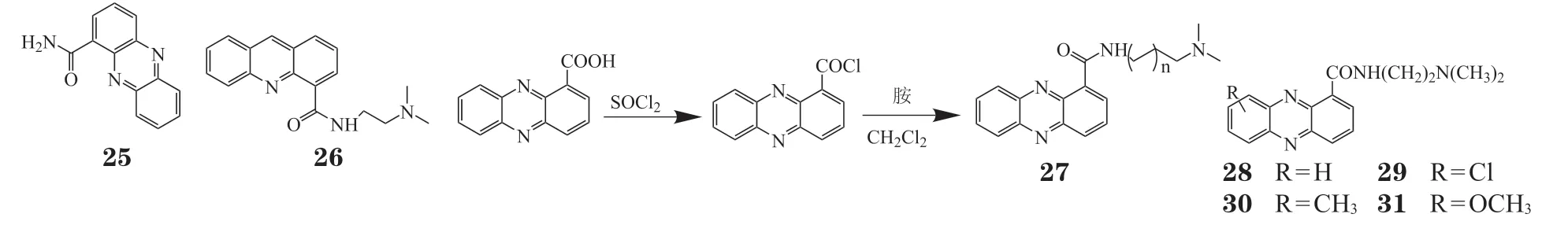
图1 吩嗪-1-甲酰胺类似物的合成的过程Figure 1 Progress of synthesis of phenazine-1-formamide analogs
研究发现,8,9-苯并吩嗪类似物(32)对P388细胞的IC50为37 nmol · L-1,其活性是3,4-和6,7-苯并吩嗪类似物的10倍[33-34]。Vicker等[35]合成了一系列具有拓扑异构酶Ⅰ/Ⅱ双重抑制作用的8,9-苯并[α]-吩嗪甲酰胺类化合物(见图2),其中XR11576(33)不仅具有优异的体内外抗肿瘤活性,而且具有口服生物利用率高、药物代谢状态理想的特点,在2004年进入Ⅰ期临床试验[36]。进一步研究表明,在这些化合物的甲酰胺侧链上引入手性基团可提高对映体的活性,如化合物33对H69/P、H69/LX4、L23/Pd和L23/Rd细胞的IC50分别为23、29、10和12 nmol · L-1,比化合物34的活性更优。
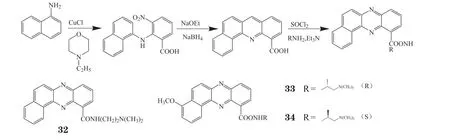
图2 苯并[α]-吩嗪甲酰胺类化合物的合成过程Figure 2 Progress of synthesis of benzo [α]-phenazine formamide compounds
Wang等[37]从化合物32为出发,合成了一系列多取代吩嗪酰胺类化合物(见图3),其中化合物35对H69/P细胞具有增殖抑制活性,IC50为0.104 μmol · L-1。化合物36对H69/LX4细胞的IC50为0.198 μmol · L-1。
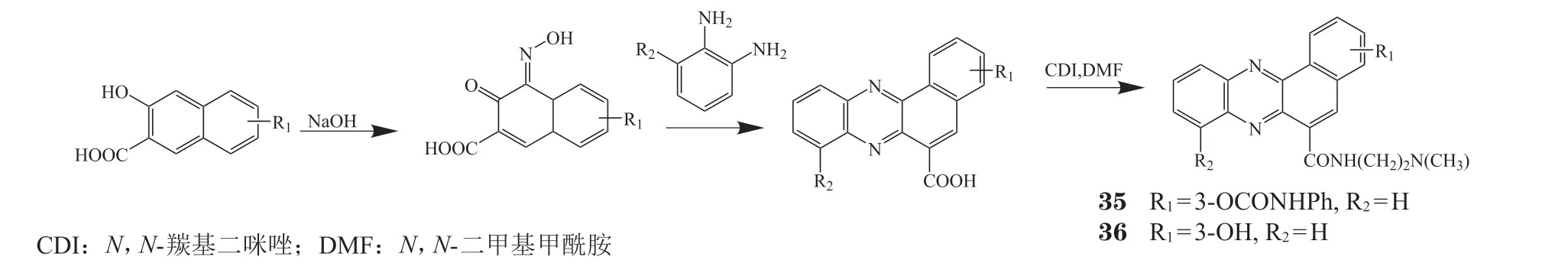
图3 苯并吩嗪-6-酰胺类化合物的合成过程Figure 3 Progress of synthesis of benzophenazine-6-amide compounds
化合物37和38是吩嗪环上含有不同取代基的8,9-苯并稠环化合物。化合物37与DNA的结合具有非特异性,其与DNA的相互作用方式取决于药物浓度:在低浓度时,以插层结合为主;在高浓度时,以静电结合为主。化合物38是拓扑异构酶Ⅱ抑制剂,具有广谱的抗肿瘤活性和抑制肿瘤转移的作用[38]。2002年Samata等[39]发现化合物38还能通过干扰胸苷激酶mRNA的表达来抑制肿瘤生长,其对长春新碱耐药的P388白血病小鼠具有很好的疗效,20世纪90年代初在日本进入Ⅱ期临床试验,但由于其不良反应而被终止试验[40]。
2.2 二聚体吩嗪
2000年,Spicer等[41]设计与合成了具有拓扑异构酶Ⅰ/Ⅱ双重抑制活性的双吩嗪-1-甲酰胺类化合物(见图4)。构效关系研究表明,当9位用亲脂性基团如CH3、Cl取代时,得到化合物39和40其对酶抑制活性显著提高。化合物39和40对P388细胞的IC50分别为15和39 nmol · L-1。
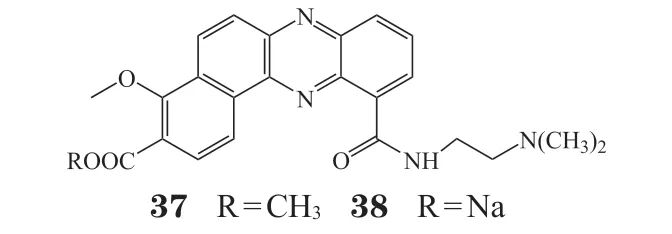
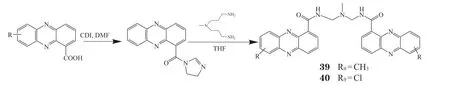
图4 双吩嗪-1-甲酰胺化合物的合成Figure 4 Synthesis of diphenazine-1-formamide
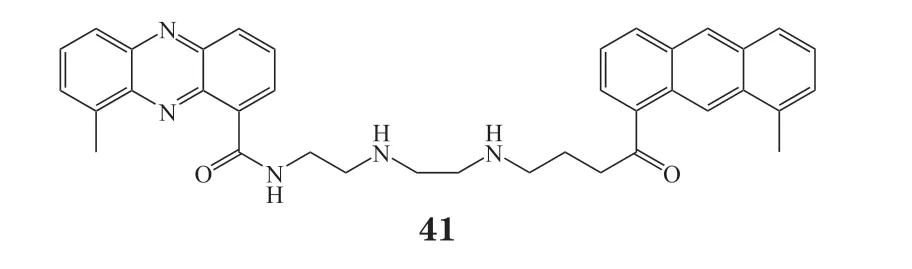
XR5944(41)是一种新型的双吩嗪化合物,对人和小鼠肿瘤细胞表现出优异的体内外增殖抑制活性,已进入Ⅰ期临床试验。Byers等[42]发现化合物41通过选择性抑制细胞RNA聚合酶的转录来发挥抗肿瘤作用,其对P388、Lewis和Jurkat细胞的IC50分别为21、3和0.2 nmol · L-1。但该化合物的细胞毒性和pKa之间缺乏相关性,因此很难研究其最佳剂量[43]。2007年,Li等[27]报道了化合物41能结合雌激素反应元件(estrogen response element,ERE)序列来抑制雌激素α受体活性,这种新作用机制可能有助于克服目前抗雌激素治疗的耐药性。2020年Serobian等[44]深入研究了化合物41的结构变化与其生物学特性之间的关系。
2.3 氮氧化吩嗪
由于肿瘤细胞生长过于迅速,无法从血液中获取足够的氧气供给生长,所以呈现出一种缺氧状态。替拉扎明(tirapazamine,42)在缺氧条件下对HCT-116细胞具有较强的增殖抑制活性,IC50为1.77 μmol · L-1[45]。2005年,Cerecetto等[46]以 苯 并 呋 喃与苯酚衍生物通过杂环扩环反应,合成了结构类似于化合物42的一系列吩嗪氮氧化物(见图5),并测定了缺氧和有氧条件下对V79细胞的细胞毒性,其中化合物43、44和45被证实在缺氧状态下触发细胞凋亡[47]。Pachon等[48]报道含有可还原的硝基和亲电性的氯乙酰胺基团的化合物46对Caco-2细胞具有增殖抑制活性(IC50= 4.8 μmol · L),其通过caspase-9和细胞色素c通路诱导G2/M细胞周期阻滞而导致细胞凋亡。当化合物46的硝基被活性较低且不可还原1,3-二 唑取代,得到的化合物47的细胞毒性明显减弱[48]。
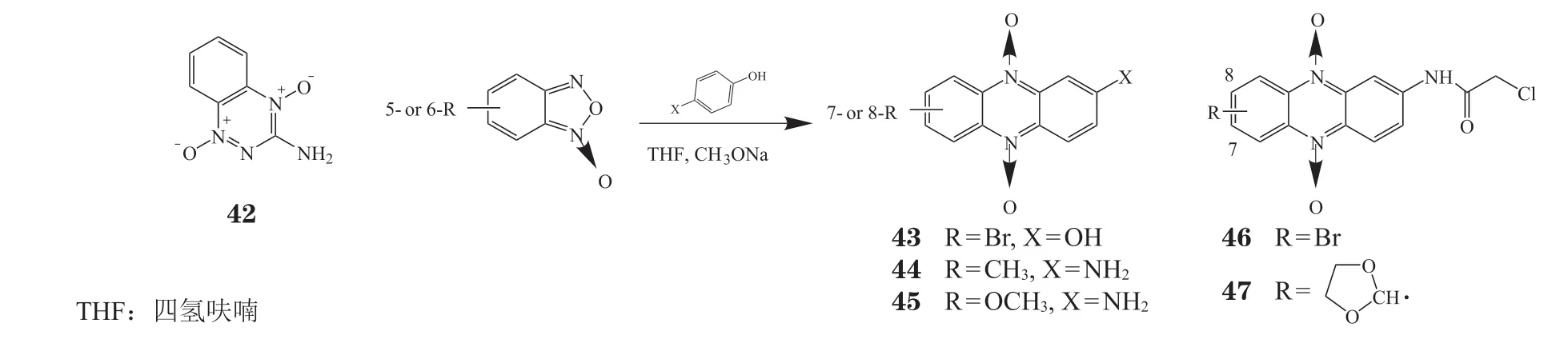
图5 替拉扎明和吩嗪氮氧化物的合成过程Figure 5 Progress of synthesis of telazamine and phenazine nitrogen oxide
2017年,Viktorsson等[49]以1,6-二羟基吩嗪的合成为关键步骤,实现了吩嗪5,10-二氧化物iodinin(48)和myxin(49)及其衍生物的高效全合成(见图6),其在有氧和缺氧条件下对MOLM-13细胞有较强的增殖抑制活性。化合物48对AML和APL细胞均具有高选择性细胞毒性[50-51]。在有氧和缺氧条件下,49对HCT-116细胞的IC50分别为1.75和4.2 μmol · L-1[45]。机制研究表明化合物48和49与DNA的C-G碱基对发生相互作用,从而抑制DNA模板控制的RNA合成。
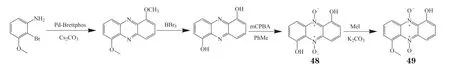
图6 氮氧化吩嗪iodinin和myxin的合成过程Figure 6 Progress of synthesis of phenazine Indole and Myxin nitroxide
2.4 吩嗪金属配合物
目前,新型光敏剂(photosensitizer,PSs)在光动力疗法(photo dynamic therapy,PDT)中的应用发展迅速,尽管PSs能很好地靶向DNA,但对正常细胞选择性差。Maity等[52]合成了吩嗪的铂配合物50,在365 nm的长波紫外线照射下,其细胞毒作用明显增强。用苯基或二茂铁基炔配体取代化合物50中的氯配体,得到的配合物51和52的暗毒性减弱,但在UV-A光照射下对HeLa和MCF-7的细胞毒性明显增强。化合物52比51的活性低,表明二茂铁基对增强配合物的光细胞毒性具有促进作用[52]。
以吖啶基双吡啶吩嗪为配体的光敏剂53在可见光下对HeLa和MCF-7细胞有明显的光毒性(IC50<0.6 μmol · L-1),在光照和黑暗条件下,其对正常细胞HPLlD无毒[53]。Rochford等[54]报道了4种铜配合物对MCF-7和SKOV-3细胞具有细胞毒性,毒性由强至弱分别为化合物54、55、56、57。
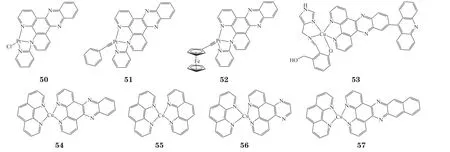
Alsaedi等[55]研究了联吡啶配体的铜(Ⅰ)配合物与DNA的结合及细胞毒性,评价了在二吡啶[3,2-a:2',3'-c]并吩嗪(DPPZ)配体上结合不同官能团对肿瘤循环DNA(circulating tumor DNA,CTDNA)结合的影响。研究结果表明,官能团影响了结合模式,从而影响了结合亲和力的强度。不同官能团与DNA结合后相互作用的位点和方式不同。DPPZ配体上的官能团对抗癌活性具有重要作用。在5种铜配合物中,含氰配合物(60)的抗癌活性最好,对M-14细胞的IC50为9.14 μmol · L-1,对MCF-7的IC50为7.33 μmol · L-1。
钌配合物因其抗肿瘤活性高、毒性低被认为是治疗肿瘤的优良药物。目前,2种钌(Ⅱ)配合物NAMI-A和KP1019已进入Ⅱ期临床试验[56]。Zhao等[57]通过引入不同的取代基来调节吩嗪配体的电子云密度,设计了一系列聚吡啶Ru(Ⅱ)-DPPZ吩嗪类钌配合物。这类配合物对一些肿瘤细胞有很好的抑制作用,同时还能抑制肿瘤的体外迁移和侵袭,尤其是Ru(Bpy)2BEDPPZ(63)表现出比其他配合物更好的抗肿瘤活性,对三阴性乳腺癌细胞MDAMB-231的IC50为17.2 μmol · L-1,优于顺铂(IC50= 20.9 μmol · L-1)。这可能由于吩嗪-钌配合物具有较大的芳香平面结构,与顺铂相比能更好地与DNA结合。构效关系研究表明,配体中芳香族平面环数目的增加能有效提升抗肿瘤活性[57-58]。
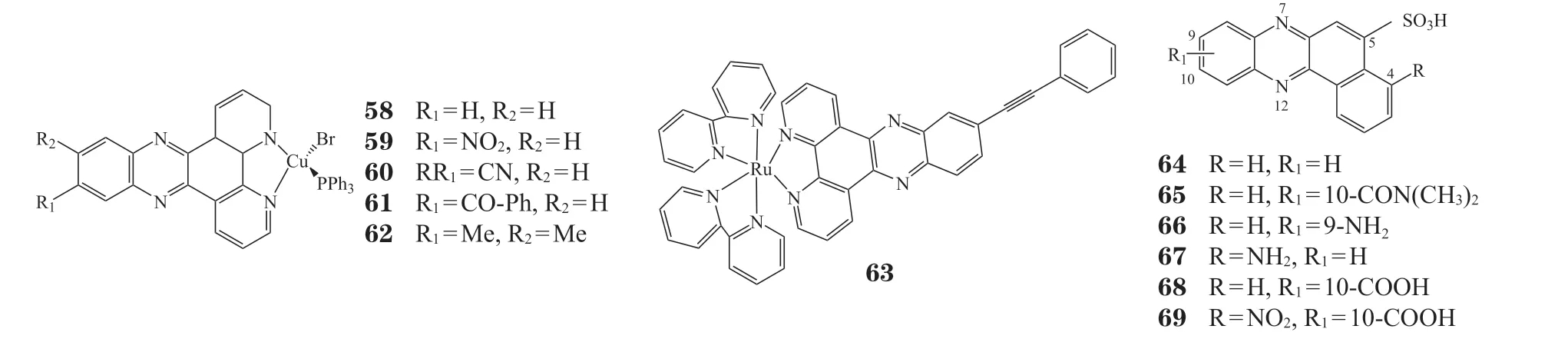
2.5 苯并吩嗪-5-磺酸及其类似物
苯并吩嗪-5-磺酸及其类似物的多环骨架能够嵌入 DNA,多个氮原子的存在能降低非氮多环体系的毒性。Moorthy等[59]合成了一系列苯并[a]吩嗪-5-磺酸衍生物,其中化合物65对HL-60细胞的增殖抑制活性最好,IC50为19 μmol · L-1。
2.6 其他吩嗪及其类似物
2015年,Moris等[60]合成了4个2,3,7-三取代吩嗪化合物(70~73)(见图7),其对人胰腺癌细胞 MiaPaca-2 具有细胞毒性,IC50分别为0.06、21、75和7 μmol · L-1,均优于吉西他滨(IC50>200 μmol · L-1),其中化合物70和73对耐药性MiaPaca胰腺癌细胞最有效[60]。
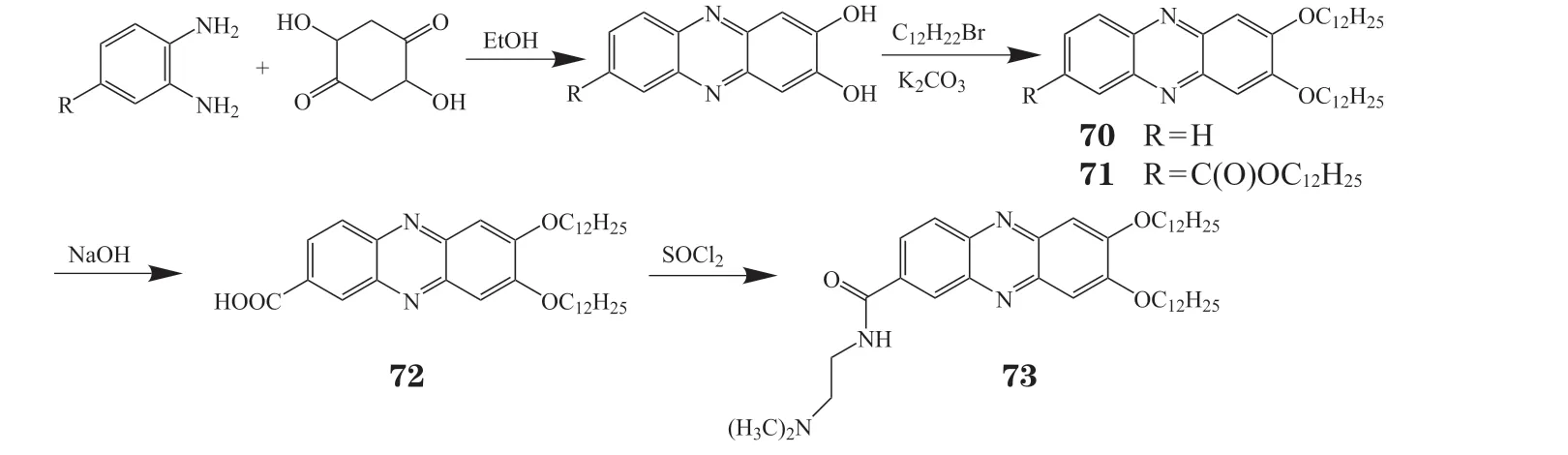
图7 2, 3, 7-三取代吩嗪化合物的合成过程Figure 7 Progress of synthesis of 2, 3, 7-trisubstituted phenazine compounds
小分子氧化应激诱导剂作为一种细胞疗法近来受到关注[61]。吩嗪甲硫酸盐化合物74作为一种促氧化剂,被广泛用作偶联还原型烟酰胺腺嘌呤二核苷酸磷酸[nicotinamide adenine dinucleotide phosphate,NAD(P)H]生成还原性四唑盐过程中电子转移的反应物。Hua等[62]首次报道化合物74对A375、G361和LOX细胞表现出较好的细胞毒性,其IC50为1 ~ 10 μmol · L-1,且能诱导致死性氧化应激和线粒体毒性应激来消除恶性黑色素瘤。
中国药科大学奚涛教授课题组从海洋放线菌次生代谢产物中分离出天然吩嗪产物NHP,该化合物对多种癌细胞表现出细胞毒性[29]。随后又设计合成了一系列新型2-氨基吩嗪衍生物(见图8)。化合物75、76、77和78在体外表现出良好的抗肿瘤活性,其中化合物75对K562和HepG2细胞具有与顺铂相当的抗癌活性,且对正常细胞293T毒性很小[63]。
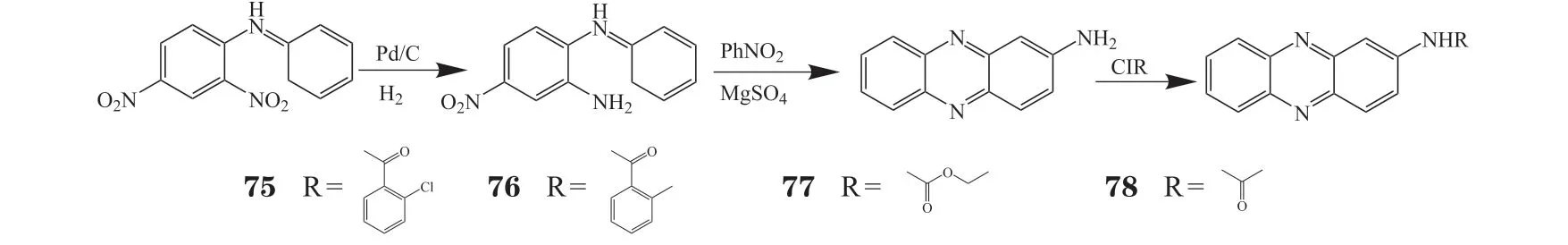
图8 2-氨基吩嗪衍生物的合成过程Figure 8 Progress of synthesis of 2-aminophenazine derivatives
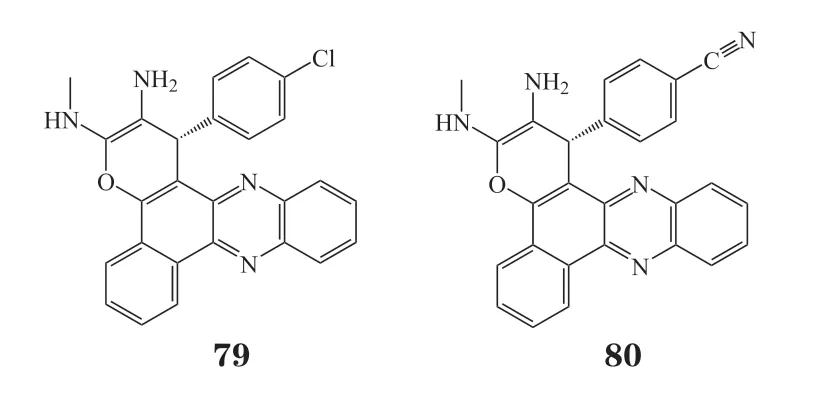
Nagaraju等[64]开发了一种新的一锅三组分反应合成苯并[α]吡喃并[2,3-c]吩嗪衍生物。该反应通过一次合成操作生成2个C-C键和1个C-O键来形成吡喃吩嗪环。反应简单,无需色谱柱纯化即可得到产品。氯和氰基取代的苯并[α]吡喃-[2,3-c]吩嗪衍生物79和80对B16-F10细胞的抑制活性最强,IC50分别为0.31和0.11 μmol · L-1。分子对接研究表明,这些合成吩嗪衍生物与细胞外调节激酶2(extracellular regulated protein kinases 2,ERK2)受体的三磷酸腺苷(adenosine triphosphate,ATP)结合位点作用发挥抗肿瘤活性。
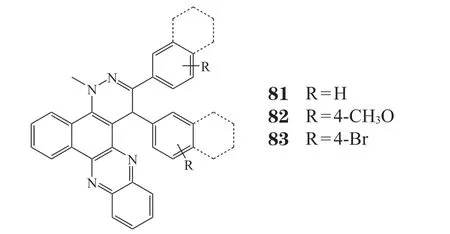
Le-Nhat-Thuy等[65]采用微波一锅法合成新型功能化苯并[α]哒嗪[3,4-c]吩嗪衍生物,并对它们的生物学特性进行评价。结果表明,化合物81、82对KB、HepG2、LU1和MCF7人肿瘤细胞具有良好的细胞毒活性。此外,化合物83对金黄色葡萄球菌和枯草芽孢杆菌有良好的抗菌活性,其IC50<6 μmol · L-1。
受抗肿瘤活性天然吩嗪的启发,笔者所在课题组基于“分子杂合”策略设计并合成了一系列苯并吩嗪并吡喃衍生物[66]、含不同取代基的吩嗪并吡喃衍生物[67]以及含有吡喃、吲哚和三氮唑的吩嗪杂合分子[68](见图9),其中最有潜力的化合物84对HepG2肝癌细胞表现出优异的体内外增殖抑制活性[67]。笔者所在课题组基于酶反应、免疫荧光和非还原性Western blot实验,进一步证实化合物84的作用靶点为Ⅰ型硫氧还蛋白还原酶(thioredoxin reductase 1,TrxR1)[69]。最近,笔者所在课题组构建了一系列含有迈克尔受体药效团的吩嗪杂合分子,其中化合物85对Bel-7402细胞具有较好的抗增殖活性,而对L02细胞无毒,表现出一定的选择性和安全性,且证实该化合物通过抑制TrxR1触发氧化应激从而破坏硫氧还蛋白系统,诱导凋亡信号调节激酶-1发挥作用导致细胞凋亡[73]。
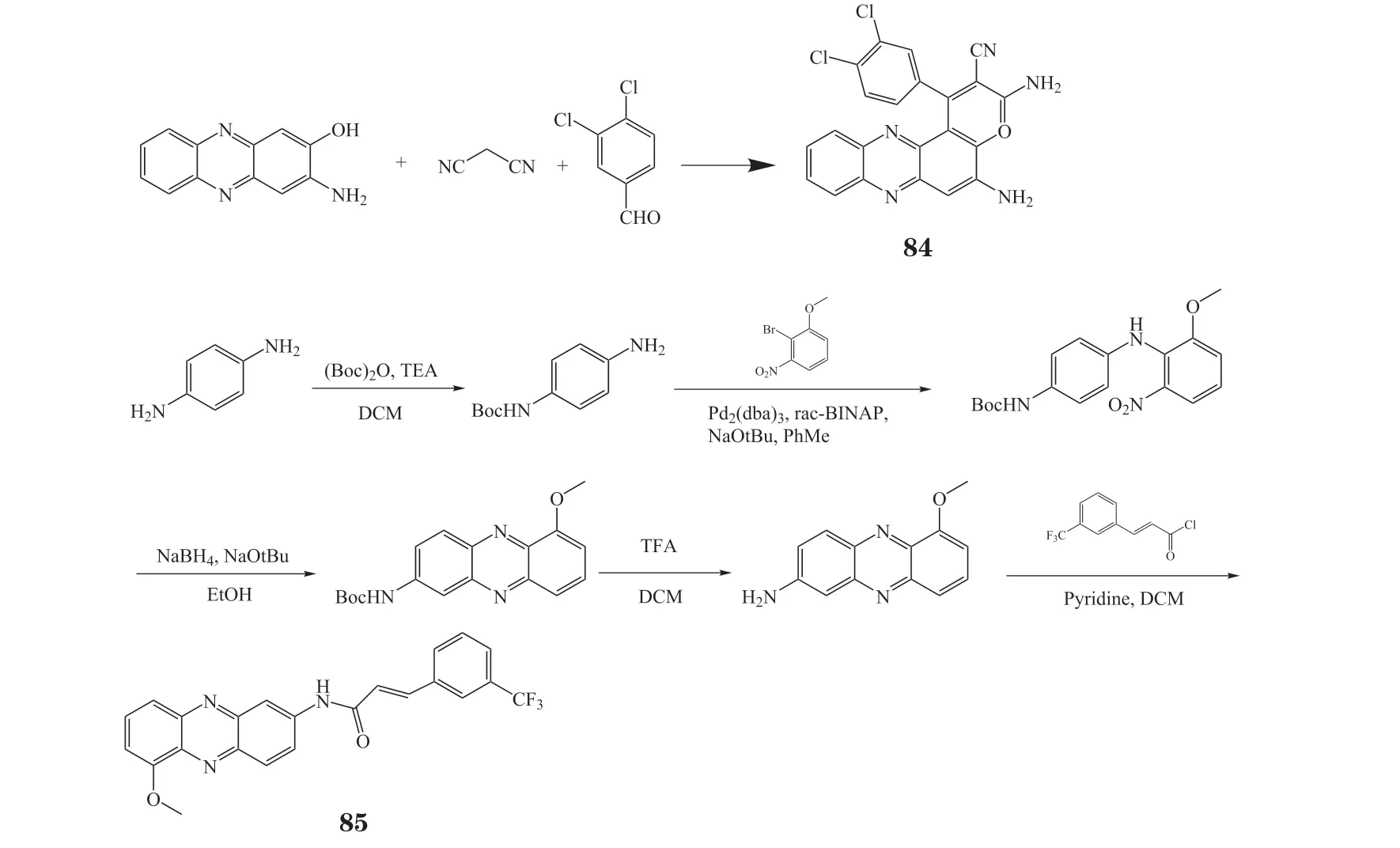
图9 化合物84和85的合成过程Figure 9 Progress of synthesis of compounds 84 and 85
3 作用机制
3.1 DNA嵌插剂
DNA嵌插剂是一类能与DNA相互作用并与之形成复合物的多环芳香化合物,可直接抑制DNA的复制与转录,或进一步活化裂解DNA而对其结构和功能产生影响。具有平面结构的芳香性吩嗪与DNA碱基对之间的π键相互作用导致插层,具体表现为UV/vis光谱吸光度降低和波长红移。苯并[α]吩嗪-5-磺酸类似物64是典型的DNA嵌插剂[59],其对HL-60细胞的增殖抑制活性比顺铂更优。前面提及的化合物48和49同样是与各种多核苷酸发生相互作用[50,70],其主要通过DNA嵌入、与RNA聚合酶结合,或与核糖核苷5'-三磷酸结合来抑制DNA模板控制的RNA合成。化合物25与DNA的结合没有表现出任何碱基特异性,而化合物49与GC碱基对富集区能产生强静电相互作用。
3.2 对拓朴异构酶Ⅰ/Ⅱ抑制作用
拓扑异构酶是存在于细胞核内的一类酶,它们通过催化DNA链的断裂和结合,从而控制DNA的拓扑状态。哺乳动物体内主要存在两种拓扑异构酶:DNA拓扑异构酶Ⅰ(TopⅠ)通过形成短暂的单链裂解-结合循环,催化DNA的拓扑异构状态的变化;拓扑异构酶Ⅱ(TopⅡ)通过瞬间引起双链酶桥的断裂,然后打通和再封闭以改变DNA的拓扑状态。常用药物如喜树碱(TopⅠ抑制剂)、依托泊苷和阿霉素(TopⅡ抑制剂)已被广泛用于癌症化疗[35]。吩嗪-1-甲酰胺衍生物26和33被发现对TopⅠ和TopⅡ具有双重抑制活性。化合物38对TopⅠ的抑制作用较弱,但对TopⅡ具有强抑制作用,其通过干扰TopⅡ的DNA链传递活性产生细胞毒性[71]。
3.3 对硫氧还蛋白还原酶抑制作用
TrxR是一种二聚体黄酮蛋白,具有暴露于蛋白表面且高度亲核的碳末端Sec 498残基。哺乳动物细胞主要含有3种亚型的TrxR,即胞质蛋白TrxR1(分布于细胞质)、线粒体蛋白TrxR2(分布于线粒体)和睾丸特定硫氧还蛋白谷胱甘肽还原酶TrxR3(分布于睾丸组织)。TrxR可以调节机体的氧化还原反应,促进细胞的生长和繁殖。大量临床观察和实验研究表明TrxR的过度激活和功能障碍与许多疾病的产生与发展有着密切的联系,例如癌症和神经退行性疾病[72]。由于多种肿瘤细胞对TrxR高表达,近年来TrxR被认为是具有极大研究价值和开发潜力的抗肿瘤靶标。如前所述,笔者所在课题组最近合成的吩嗪类似物84和85就是一类结构新颖的TrxR1抑制剂[69,73],为吩嗪类抗肿瘤药物的开发提供了新的思路。
4 结语与展望
本文总结了近年来天然来源的吩嗪及其衍生物和化学合成吩嗪类似物的结构、抗肿瘤活性和作用机制的研究进展。尽管大量抗肿瘤吩嗪天然产物已被分离得到,但研究人员对其构效关系和作用机制的理解仍不够透彻,缺乏对其生物化学性质的深入认识。人工合成吩嗪类似物可在吩嗪母核不同位置引入取代基或官能团,进而深入研究其结构与活性之间的密切关系。然而,结构多样性吩嗪杂环的合成通常面临原料不易得、反应条件苛刻、产率低、反应步骤多等缺点,使该类化合物的活性筛选和生物评价受到限制。探寻简单高效、模块化的多样性吩嗪成环方法,是未来需要迫切解决的重要问题之一。由于具有平面结构的吩嗪分子与DNA的结合缺乏选择性,容易产生普遍的细胞毒性,在今后的研究中,围绕吩嗪母核进行定向修饰和功能化,比如利用药效团拼接或分子杂合技术,使化合物能特异性结合靶点蛋白发挥药效,也是今后需要突破的方向之一。另外,基于吩嗪自身荧光特性开发针对特定蛋白的分子探针,对研究吩嗪类化合物的生物功能具有重要意义,有望获得具有“诊疗一体”功能的活性分子[74]。总的来说,基于吩嗪及其类似物重要的药学研究价值,可以预见该类化合物在医药研发领域必将具有广阔的发展前景。
