基于聚苯胺纤维素纳米纤维的磁组装电极电化学氧化处理酸性红G废水研究
2022-10-13杨长安赵伟鹏
杨长安, 赵伟鹏, 邵 丹
(陕西科技大学 材料科学与工程学院, 陕西 西安 710021)
0 引言
我国人均淡水资源低于世界平均水平,但随着经济发展,工业用水量逐年增加.工业废水的治理与回用是缓解水资源压力的关键一环,受到国家的高度重视.主流工业废水处理方法包括物理法、化学法、生物法[1]等,但这些方法处理难降解有机废水时均存在各自的短板[2].所谓难降解有机废水是指污染物化学稳定性高或毒性强的有机废水,难以生化降解[3].电化学氧化法是高级氧化法的一种,其具有清洁、高效、灵活等特点,是一种新型废水处理技术[4].阳极材料是电化学氧化法的核心,典型如采用Ti/PbO2、Ti/Sb-SnO2或Ti/Ti4O7等高析氧电位阳极电解处理废水的过程中,阳极表面能够高效产生并积累具有强氧化性的物理吸附羟基自由基,其对难降解有机污染物及其降解中间产物的氧化效果较好[5].因此,电化学氧化法适合处理难降解有机废水,可作为以生物法为典型的主流方法的重要补充.
电化学氧化难降解有机废水的关键问题是污染物降解电流效率低和伴生的高能耗问题.由于阳极有机物降解反应与析氧反应为竞争反应,在处理低浓度废水时(尤其是低于100 ppm时),污染物向阳极的传质较差,降解反应受传质控制,因此导致副反应显著,降解电流效率降低,能耗升高[6].如能将低浓度有机污染物在阳极附近进行浓缩富集,则能有效改善传质问题、提高处理电流效率并降低处理能耗.
近年来,随着阳极材料的发展,出现了一种在形式上介于传统二维电极和三维电极之间的新型阳极形式,即磁组装电极(Magnetically Assembled Electrode,MAE).MAE本质是一种模块化电极,其利用磁铁将具有磁性的副电极材料吸附于二维主电极上,引入颗粒后相比于主电极来说体积变化不大,但是大大增加了与降解介质的接触面积,即赋予MAE较大的面积体积比及大量的催化活性位点,并使MAE具有多样性、性能灵活可调性以及良好的可回收性[7-9].MAE的表面组成和结构可根据废水成分变化进行相应的调整,因此当废水成分波动时,基于MAE的电化学水处理系统无须停车更换电极即可实现动态废水的全时高效处理.
将磁性四氧化三铁(Fe3O4)、导电高分子聚苯胺(PANI)及多孔纤维素纳米纤维(CNFs)气凝胶三种材料进行复合所得到的纤维素纳米纤维/聚苯胺磁性气凝胶(CNFs/PANI)不仅可实现电性能、吸附性能和磁性能的复合,而且还具有良好的机械性能和为大孔/介孔共存的多级孔结构,既有利于污染物的传输,也便于污染物的吸附[10].因此,本研究拟采用这种新型磁性复合碳基吸附材料作为MAE中的副电极,以加强废水中有机污染物向电极表面的迁移与富集,实现更为高效的有机废水处理.从图1可以看出,本研究设计的新型磁组装电极由三部分构成,内侧为磁铁,中间为Ti/PbO2主电极,外层为CNFs/PANI副电极.从图1可以看出,本研究设计的新型磁组装电极由三部分构成,内侧为磁铁,中间为Ti/PbO2主电极,外层为CNFs/PANI副电极.本研究欲综合Ti/PbO2的优势(电催化活性高,导电性好,阳极尺寸稳定,工作寿命长,成本低)纤维素纳米纤维的优点(高机械强度、高吸附通量)及聚苯胺的特点(良好的电活性与稳定性、对有机污染物吸附作用强),以期获得综合性能优良的新型MAE.

图1 新型磁组装电极的结构及强化传质功能示意图
1 实验部分
1.1 电极制备与组装
Ti/PbO2主电极的制备方法遵循课题组以往惯例[11-12].简而言之,在预处理后的Ti片上采用阴极电沉积-热氧化法负载Sb-SnO2中间层,然后采用阳极电沉积法负载PbO2层.磁性CNFs/PANI的制备按照东华大学吕伟等[8]的方法进行.
将制备好的磁性CNFs/PANI成品块体材料裁剪为5 mm厚的薄片,后采用NdFeB磁铁将CNFs/PANI薄片吸引并完全覆盖在Ti/PbO2表面,即为MAE,此新型MAE命名为PbO2/Fiber.
1.2 电极表征
采用扫描电子显微镜(Gemini SEM 500 Field Emission Scanning Electron Microscope)观察CNFs/PANI块体材料结构与形貌.
采用三电极体系电解池对PbO2/Fiber(主电极有效面积1 cm2)进行各项电化学表征(循环伏安测试,线性扫描测试).电化学测试仪器为CS310H型电化学工作站,测试采用三电极化学测量体系.以电化学工作站作为电源,所制备的电极作为工作电极.以与制备电极尺寸相差不大的铜片作为对电极,和工作电极的间距保持在0.5 cm.以饱和甘汞电极(SCE:0.241 V (vs.SHE))为参比电极,紧贴工作电极,置于距离被测试的电极有效区域相近的一侧.电位扫描速率为0.05 V·s-1.
1.3 电化学氧化降解
采用两电极体系电解池对200 ppm酸性红G溶液(250 mL)模拟染料废水和造纸废水模拟废水进行电化学氧化降解实验对电极的催化活性进行评价.采用恒流直流电源,将PbO2/Fiber(主电极有效面积9 cm2)作为阳极,相同面积铜片作为阴极,阳极电流0.09 A.降解时间4 h,每隔1 h从溶液中取样10 mL,利用紫外可见分光光度计对样品进行紫外可见吸收光谱测试.
2 结果与讨论
2.1 副电极形貌
图2为制备的CNFs/PANI副电极的副电极电极形貌与结构表征图.图2(a)为50倍率下的电极扫描电镜图,形状与海绵相似,整体呈现为疏松多孔结构,孔的大小约为0.1 mm;图2(b)、(c)为高倍率下的电极扫描电镜图,在高倍率下的CNFs/PANI副电极依旧为疏松多孔结构,由图2可知孔径大小约在10~50nm之间;图2(d)为CNFs/PANI的吸附-脱附与粒径分布图.通过分析测试结果,判断CNFs/PANI的测试曲线属于Ⅳ型,介孔回滞环属于H1型,可以判断属于介孔材料.
在预期设想中,CNFs/PANI副电极的三维多孔结构扮演着关键作用.所获得的CNFs/PANI副电极材料确实具有三维多孔结构,其不仅具有疏松的大孔结构,而且大孔内还含有大量的介孔.


图2 CNFs/PANI电极形貌与结构表征
2.2 电极溶液界面状态和活性位点
图3为磁组装电极PbO2/Fiber在不同溶液中及不同扫速下的小范围循环伏安曲线.磁组装电极比Ti/PbO2具有更大的充放电电流,即具有更大的表面电容,说明磁组装电极的表面状态更为粗糙.此外,在图3(b)中可以发现,正向扫描时在电位0.85 V时出现明显的氧化峰,证明负载的CNFs/PANI副电极的电活性发挥作用.在硫酸钠溶液中添加酸性红G后,磁组装电极的相应电流的增加幅度较大(如图3(b)所示),说明在这个电位区间,吸附在CNFs/PANI副电极上的酸性红G分子对电极电容的贡献较大,即直接的电荷转移倾向较大.

图3 磁组装电极PbO2/Fiber在不同溶液中及不同扫速下的小范围循环伏安曲线
图4所示内容为伏安电荷点.在扫速很低时,可以近似认为所获得的伏安电荷值(qT)对应于电极表面的总活性位点数量,而扫速较高时,可以近似认为所获得的伏安电荷值(q)对应于电极外表面的活性位点数量.两种伏安电荷值的差值即为qi,对应于难以接近的内表面活性位点数量.由图4可知,Ti/PbO2电极的qT、qi与q值均显著低于磁组装电极,说明副电极的负载为PbO2/Fiber磁组装电极带来了大量的额外活性位点.在溶液中加入酸性红G后,可以发现两种电极的伏安电荷值都显著上升,尤其是对于Ti/PbO2电极,提升幅度高达约3倍,说明酸性红G在析氧电位以前就与PbO2表面的活性位点间发生了明显的电荷转移,这与上述小范围循环伏安曲线中充放电电流分析的结论是一致的.
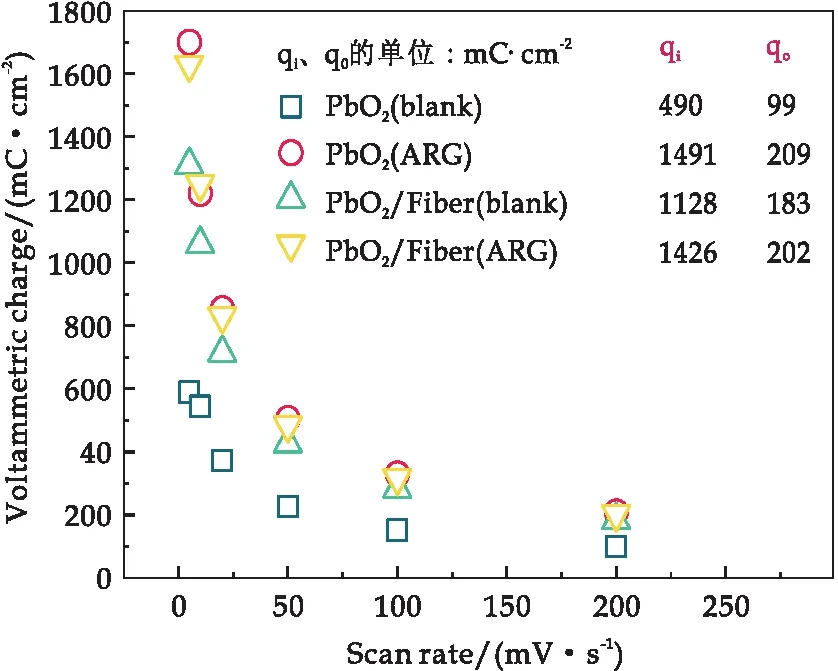
图4 磁组装电极PbO2/Fiber与对照电极PbO2在不同溶液中及不同扫速下的伏安电荷、qi与q值
2.3 电极电催化活性
图5为磁组装电极PbO2/Fiber与对照电极PbO2在不同溶液中的Tafel曲线,没有负载副电极的Ti/PbO2电极在硫酸钠溶液中对应于0.01 A电流析氧电位为1.74 V,而负载CNFs/PANI副电极后电极的析氧电位降低至1.68 V,说明副电极的引入提高了电极的析氧电催化活性,与之前电极溶液界面状态与活性位点的分析结论是一致的.溶液中加入酸性红G后,Tafel曲线位置下降,Ti/PbO2电极下降的幅度更大,说明酸性红G在两种电极表面都发生了一定程度的直接氧化,即吸附在电极表面直接失去电子,其中在Ti/PbO2电极表面发生直接氧化的倾向更为明显.

图5 磁组装电极PbO2/Fiber与对照电极PbO2在不同溶液中的Tafel曲线
图6为磁组装电极PbO2/Fiber与对照电极在不同溶液中的常规循环伏安曲线,未负载CNFs/PANI的空白对照电极其CV曲线与其余三种电极显示出了较为明显的差异.在0.5 mol/L Na2SO4溶液中,空白对照电极PbO2的最大响应电流为0.087 3 A·cm-2,其余三组的最大响应电流在0.112 A·cm-2左右,相差不大.说明电极在负载CNFs/PANI后,最大响应电流密度增高,可能是由于副电极的引入,大大增加了电极表面的总活性位点(CNFs/PANI副电极上具有的活性位点、PbO2层上的活性位点、两种不同活性位点相互接近后产生的新界面上的活性位点);随着酸性红G溶液的引入,酸性红G在电极表面发生直接氧化,且与析氧反应相互竞争,其余三组的最大响应电流变化不大的原因可能是酸性红G直接氧化的速率与析氧速率近似.

图6 磁组装电极PbO2/Fiber与对照电极在不同溶液中的常规循环伏安曲线(扫描速度0.05 V·s-1)
2.4 酸性红G的降解
图7所示内容为两种电极降解酸性红G过程中溶液样品的紫外可见吸收光谱,在电化学氧化过程中酸性红G的偶氮吸收峰(506 nm处)与苯环、萘环吸收带(300至400 nm处)都显著降低,说明电化学氧化不仅能够有效破坏酸性红G的偶氮官能团,将其分子打碎,同时也能令顽固的苯环和萘环发生开环,进一步提高其可生化性.
由图7(a)可以看出,酸性红G在未降解时,萘环吸收带的吸光度为1.413,降解1 h后吸光度下降为1.16,酸性红G在未降解时在偶氮吸收峰的吸光度为1.858,降解1 h时下降为1.048,吸光度变化较大可达0.81.而在降解2 h和3 h后,从图中可明显看出吸光度下降程度大幅度减小,一小时内偶氮吸收峰的吸光度下降约为0.31.
由图7(b)可以看出,酸性红G在未降解时萘环吸收带的吸光度下降情况与图7(a)类似,但是偶氮吸收峰的变化情况存在差异.酸性红G在降解0 h、1 h、2 h和3 h时,偶氮吸收峰的吸光度分别为1.683、1.303、0.861和0.554,基本呈线性趋势下降,但是在降解最初的1 h内,对照 PbO2电极降解效果优于磁组装电极PbO2/Fiber.综上所述,在酸性红G浓度较高时,对照 PbO2电极在偶氮吸收峰具有明显的选择性,而随着实验不断进行,酸性红G浓度不断变低的同时,对照 PbO2电极略显乏力,反观磁组装电极PbO2/Fiber对于偶氮键的降解保持稳定.
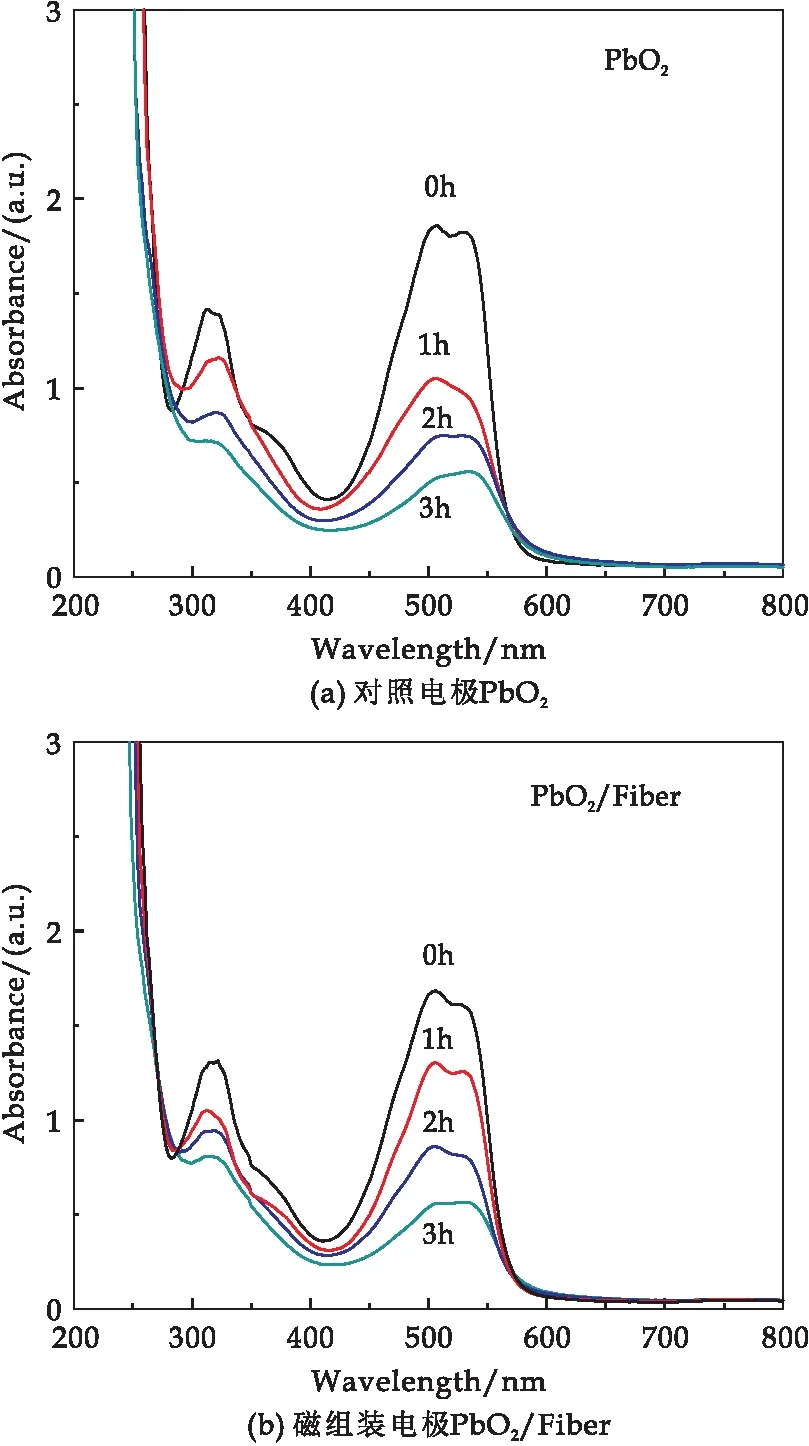
图7 磁组装电极PbO2/Fiber与对照电极PbO2降解酸性红G过程中的溶液紫外可见吸收光谱变化
图8所示内容为两种电极降解酸性红G过程中溶液样品的紫外可见吸收光谱中506 nm处吸光度变化值降低率折算为酸性红G的降解率.可以明显看出,磁组装电极PbO2/Fiber的酸性红G降解率随时间推移基本呈直线上升,而对照组Ti/PbO2的酸性红G降解率随时间推移呈曲线上升,上升速率逐渐降低,呈典型的准一级反应动力学特征,说明有机物浓度逐渐下降会严重影响降解速率,反应从始至终都是受传质控制.因此,在降解中后期(2至4 h),磁组装电极活性位点多、表面粗糙分级多孔、吸附能力强的优势逐渐显露出来,最终反超对照组电极,取得近85%的酸性红G降解效率.以上结果证明实验预期效果最终得以实现,通过CNFs/PANI副电极负载,吸附与传质加强后的有机物电化学氧化效率确实得以提高.而在降解初期(0至2 h)磁组装电极对ARG的降解率较为一般,说明降解有机物的主要力量仍是集中于主电极,副电极对主电极的遮挡一定程度上影响了初期有机物浓度大、传质情况本就较好时的降解效率,与图7所得出的结论一致,这为今后此类电极的继续优化提供了宝贵参考.
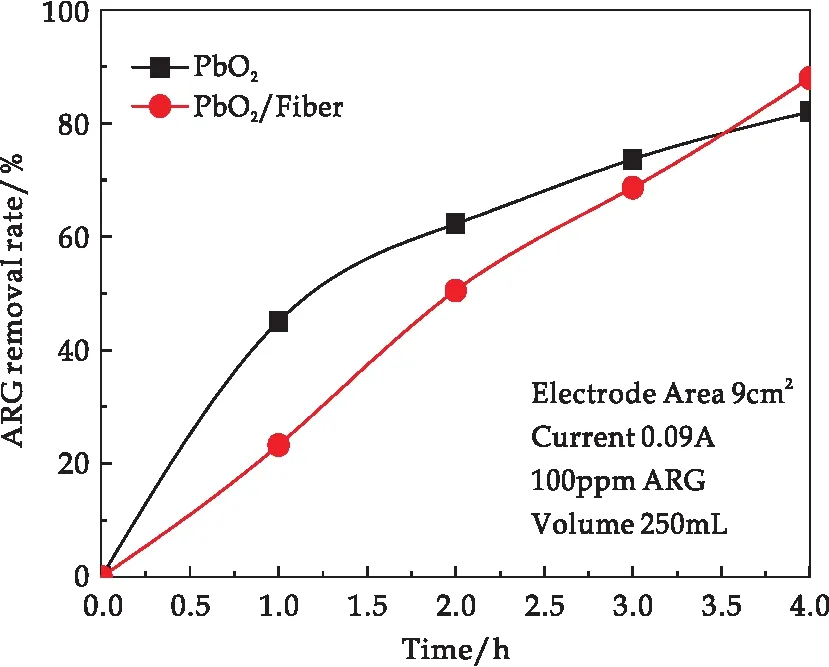
图8 磁组装电极PbO2/Fiber与对照电极PbO2的酸性红G降解率随时间变化
3 结论
在本文研究工作中,成功组装了以Ti/PbO2为主电极,以磁性CNFs/PANI分级多孔复合材料为副电极的新型磁组装电极PbO2/Fiber.相比与对照组传统Ti/PbO2电极,这种新型电极具有较多的催化活性位点与较高的电催化活性,其表面副电极的分级多孔结构有利于有机物的吸附与传质,最终使得电化学氧化降解有机物反应克服了传质控制限制,在有机物浓度较低时也能保持稳定的降解效率.
