心脏非成纤维细胞的旁分泌信号在调控心肌纤维化中的研究进展*
2022-10-13周宙张科科王震王咏刘杨
周宙, 张科科, 王震, 王咏, 刘杨△
心脏非成纤维细胞的旁分泌信号在调控心肌纤维化中的研究进展*
周宙1, 张科科1, 王震2, 王咏2, 刘杨2△
(1山东中医药大学第一临床医学院,山东 济南 250000;2山东中医药大学附属医院心内科,山东 济南 250000)
心肌纤维化;心脏非成纤维细胞;旁分泌
作为一种能够危及生命的疾病,心力衰竭当前在全球范围内已经威胁到大约6 400万人民的健康,并且患病率仍在逐年增加[1]。心肌纤维化是心力衰竭的重要病理特征,也是引起心功能不全的主要原因之一。在心肌损伤后(如过度的机械牵拉、缺血、梗死和再灌注损伤等),成纤维细胞被激活合成大量的胶原蛋白,损伤或死亡的心肌细胞被富含胶原蛋白的疤痕包裹甚至代替,在一定范围内的纤维化是心脏对原发性坏死性损伤的修复,具有保护心脏的积极作用[2]。然而,过度的胶原蛋白富集能够增加心室壁的硬度,阻碍了正常的收缩和舒张功能,诱发或加重了心力衰竭的病理过程,因此抑制或逆转过度的心肌纤维化成为治疗心力衰竭的重要靶点[3-4]。
纤维化是细胞外基质(extracellular matrix, ECM)累积的总称,心脏成纤维细胞是表达和沉积纤维状ECM的核心细胞类型,在病理性刺激下成纤维细胞被活化向肌成纤维细胞转化,后者胶原蛋白合成能力显著增加,是心肌纤维化过程中细胞外基质的主要来源细胞[5]。当前,成纤维细胞是改善心肌纤维化的研究重点,但越来越多的证据表明,心脏中的非成纤维细胞(心肌细胞、炎症细胞和内皮细胞等)同样在心肌纤维化的过程中发挥重要的作用[2]。在心肌纤维化的过程中,非成纤维细胞不仅能够通过分泌促成纤维细胞活化因子影响成纤维细胞的表型转化,并且能够分泌细胞外基质蛋白酶以启动ECM的过度重塑从而加重心肌纤维化的进程[6-7]。
心脏微环境中各类细胞的交流是通过细胞间的信号传导实现的。不同于依赖于循环系统的内分泌途径和具有靶向局限性的神经分泌途径,旁分泌作为第三种常见细胞间信号因子的交流途径是心肌微环境内各细胞发生信号交流的主要形式。旁分泌信号主要通过直接分泌可溶性因子、细胞外囊泡包裹和外泌体携带等3种方式从细胞向胞外释放[8]。纤维化过程中,邻近的非成纤维细胞能通过以上3种旁分泌方式向组织微环境中释放信号因子,并被成纤维细胞接受,诱导成纤维细胞功能和表型发生改变,促进了心肌纤维化的产生和发展。此外,非成纤维细胞之间的相互影响也参与了心肌纤维化的调控。但是,当前仍缺乏基于旁分泌治疗心肌纤维化的有效药物。本文将就旁分泌介导的非成纤维细胞与成纤维细胞之间的相互作用及其对心肌纤维化的影响进行综述。
1 心脏非成纤维细胞的旁分泌信号对心肌纤维化的调控
1.1心肌细胞的旁分泌信号对心肌纤维化的调控心肌细胞是心脏泵血功能的效应细胞,也是心功能障碍的主要损伤单元。心肌细胞对于外界刺激十分敏感,除了发挥舒缩功能外,心肌细胞还参与了心脏微环境稳态的调控[9]。受损心肌细胞向外界释放大量的促炎因子和趋化因子,并且坏死的细胞还能触发先天性免疫通路引起强烈的炎症反应,炎症细胞(白细胞和单核细胞)的大量浸润是心肌ECM发生重塑的重要原因[10-11]。研究显示接受主动脉缩窄术后,活性氧(reactive oxygen species, ROS)介导了小鼠心肌细胞结缔组织生长因子(connective tissue growth factor, CTGF)的上调并以旁分泌形式释放,心肌细胞源的CTGF显著激活了心肌成纤维细胞,表现出大量的胶原蛋白合成[12]。此外,心肌细胞可以通过介导微小RNA(microRNA, miRNA)向胞外的释放调控心肌纤维化[13]。miRNA作为一种进化的保守机制,能够与靶基因的转录本的部分区域互补而形成RNA诱导沉默复合体,抑制其蛋白的合成[14]。Li等[15]发现,在心肌梗死或缺血再灌注状态下,心肌细胞分泌富含miR-30d的细胞外囊泡(extracellular vesicles, EVs),miR-30d通过沉默心肌成纤维细胞中的整合素α5(integrin alpha-5,),阻碍了ITGA5介导的心肌成纤维细胞活化。不仅是缺血性疾病,在机械牵拉刺激下(如心脏前负荷过高),心肌细胞与心肌成纤维细胞间也存在信号传递。接受主动脉缩窄术的心脏压力超负荷大鼠心肌存在大量纤维化病变,其病理机制包括压力诱导的心肌成纤维细胞活化和内皮-间充质转化(endothelial-mesenchymal transition, EndMT;内皮细胞转化为成纤维细胞为主的间充质细胞)等多种病理过程[16-17]。在接受主动脉缩窄术后的小鼠心肌组织中miR-378的水平显著提高,而体外实验显示心肌细胞(而不是成纤维细胞)在接受机械牵拉刺激后将携带大量miR-378的外泌体从胞内多囊泡小体释放进入心肌微环境,miR-378靶向心肌成纤维细胞中丝裂原活化蛋白激酶激酶6(mitogen-activated protein kinase kinase 6, MKK6)的mRNA而抑制其蛋白表达,并促进p38丝裂原活化蛋白激酶(mitogen-activated protein kinase, MAPK)/Smad2/3信号通路因缺少MKK6激活而失去的促成纤维细胞活化能力[18]。这可能是心肌代偿性调控心肌纤维化的机制之一。
纤维化也是心房颤动的重要病理基础[19]。降钙素(calcitonin, CT)是一种主要(但不完全)由甲状腺滤泡旁细胞产生的激素,它参与了骨吸收和胶原蛋白转换[20]。Moreira等[21]观察到CT也能够在人类心房组织中表达,并且心房的心肌细胞是CT的主要非甲状腺来源;与对照组相比,沉默的自发性房颤小鼠房颤的出现时间明显提前且心房表现出显著的心肌纤维化改变,而CT过表达组与对照组无显著差异;进一步研究显示心肌细胞通过旁分泌释放的CT能够与心房成纤维细胞表面的CT受体结合,通过上调Gαs/cAMP通路抑制骨形态发生蛋白1(bone morphogenetic protein 1, BMP1)的表达;BMP1参与了胶原前体向成熟胶原的加工过程,因此心肌细胞通过旁分泌途径分泌的CT能够抑制成纤维细胞成熟胶原的合成。上述研究结果提示CT具有延缓心房心肌纤维化改变的作用,这为基于抑制心房纤维化治疗心房颤动提供了新的靶点。
1.2心脏微血管内皮细胞的旁分泌信号对心肌纤维化的调控心脏微血管内皮细胞作为心脏脉管系统的保护屏障也是对循环系统来源刺激(缺氧、氧自由基、炎症因子、血流应剪切力异常等)的最早反应单位[22]。病理条件下,微血管内皮细胞发生EndMT,为心肌纤维化提供了额外的成纤维细胞来源,在高血压、心肌梗死和心力衰竭等疾病的心肌纤维化过程中发挥重要作用[23-25]。除此之外,内皮细胞还能通过旁分泌信号诱导邻近成纤维细胞的活化。在缺氧/复氧条件下(如缺血再灌注),心脏微血管内皮细胞被Snai1诱导发生EndMT,EndMT细胞能够向微环境中分泌大量的CTGF以诱导邻近心肌成纤维细胞的活化,这种纤维化调控可能是心脏发生再灌注损伤的原因之一[7]。Zhang等[26]通过构建多种小鼠心肌纤维化模型(主动脉缩窄、心肌梗死或血管紧张素II诱导),观察到E26转化特异性相关基因[E26 transformation-specific (ETS)-related gene,]在心肌组织中的表达显著下调;进一步研究表明,表达的降低导致内皮细胞中内皮素1(endothelin-1, ET-1)表达上调并通过旁分泌途径激活了心肌成纤维细胞的蛋白激酶B(protein kinase B, PKB/AKT)/糖原合酶激酶3β(glycogen synthase kinase 3β, GSK3β)、MAPKs和Smad等信号通路,从而诱导成纤维细胞的表型活化;在内皮细胞中特异性过表达ERG蛋白或使用ET-1抗体后,多种刺激下的心肌纤维化被显著抑制。这为从阻断微血管内皮细胞与成纤维细胞间的信号传导而抑制心肌纤维化提供了思路。值得注意的是,微血管内皮细胞在促进心肌纤维化的同时也可以通过旁分泌途径诱导心肌细胞的凋亡。在缺氧/复氧内皮细胞模型中,内皮细胞中转化生长因子β1(transforming growth factor β1, TGFβ1)显著上调,作为已知EndMT的最强诱导因子,TGFβ1在促进内皮细胞发生EndMT的同时以旁分泌形式被释放,作用于心肌细胞表面的1型TGFβ受体(TGFβ receptor type-1, TGFβR1),诱导心肌细胞凋亡,新的心肌缺失和心肌纤维化之间形成恶性循环,加重了心功能的恶化[27]。
1.3巨噬细胞的旁分泌信号对心肌纤维化的调控高炎症性和细胞外基质重塑是心肌梗死后心脏的关键病理机制,而在二者的病理过程中都存在巨噬细胞的参与[28-29]。常驻于心肌的巨噬细胞在不同病理阶段以促炎表型(M1)和抗炎表型(M2)分别参与损伤组织的清除和促进修复的过程[30]。除了直接参与细胞外基质重塑外,巨噬细胞与其他心肌细胞间也存在着旁分泌信号并对心肌纤维化进行调控。心肌梗死后,心脏中常驻巨噬细胞中基质金属蛋白酶14(matrix metalloproteinase 14, MMP14)显著上调,MMP14能够促进储存于巨噬细胞胞膜的潜伏期相关肽(latency-associated peptide, LAP)和TGFβ1形成的潜伏复合体的分解,TGFβ1以旁分泌形式进入心脏组织微环境,通过对内皮细胞Smad2的磷酸化活化诱导内皮细胞发生EndMT,最终加重了心肌纤维化[31]。此外,TGFβ1也能够通过与心肌成纤维细胞的相应受体TGFβR1结合促进心肌成纤维细胞的活化[32]。因此巨噬细胞分泌的TGFβ1可能通过促进内皮细胞EndMT过程和直接活化心肌成纤维细胞等多途径参与心肌纤维化的调控。circRNA作为一种环状闭合的非编码RNA链能够与细胞中多种miRNA结合从而抑制其功能,这种吸附性抑制miRNA的能力被称为circRNA的海绵作用[33]。有研究表明,心梗后的抗炎表型巨噬细胞(M2)能够释放大量富含CircUbe3a的小细胞外囊泡(small extracellular vesicles, SEVs),M2来源的CircUbe3a通过SEVs进入心肌成纤维细胞并靶向miR-138-5p,miR-138-5p失去了对促成纤维细胞活化基因表达的抑制,因此M2通过旁分泌CircUbe3a对miR-138-5p/信号轴的调控促进了成纤维细胞的活化和心肌纤维化[34]。
1.4粒细胞的旁分泌信号对心肌纤维化的调控不仅是巨噬细胞,同样作为感知心肌组织损伤并作出反应的白细胞,粒细胞也参与心肌纤维化的调控[35]。中性粒细胞是粒细胞的一个亚型,心肌梗死中性粒细胞中前列腺六次跨膜上皮抗原2(six-transmembrane epithelial antigen of prostate 2, Stamp2)的缺乏使细胞中核因子κB(nuclear factor-κB, NF-κB)激活,活化的NF-κB可促进中性粒细胞以旁分泌途径释放大量髓过氧化物酶(myeloperoxidase, MPO)诱导成纤维细胞发生纤维化表型转化[36]。MPO是活化成纤维细胞的中性粒细胞来源重要介质,主要通过激活成纤维细胞中的p38 MAPK信号通路诱导成纤维细胞向肌成纤维细胞转分化,从而促进心肌纤维化[37]。
研究表明,结构异常性心肌病患者经常伴有嗜酸性粒细胞增多症[38]。而在动物实验中嗜酸性粒细胞增多对于心脏重塑的促进作用也被证实,因此嗜酸性粒细胞对心肌纤维化的影响也成为了研究重点[39]。心肌梗死后,嗜酸性粒细胞依赖白细胞介素4(interleukin-4, IL-4)介导嗜酸性粒细胞阳离子蛋白(eosinophil cationic protein, ECP)高表达,ECP能够通过旁分泌途径抑制成纤维细胞的TGF-β1/Smad2/3信号通路,进而以抑制成纤维细胞活化的方式抑制心肌纤维化的进展[40]。此外,ECP还可以作用于心肌细胞,抑制心肌细胞的凋亡和细胞肥大相关信号通路的激活[41]。因此,嗜酸性粒细胞能够通过旁分泌信号从抑制成纤维细胞活化、心肌细胞凋亡和肥大等多种途径发挥抗心脏结构重塑的功能。嗜碱性粒细胞在心肌纤维化过程中的作用少有研究,有研究表明其通过旁分泌途径释放大量IL-4参与了心脏同种异体移植物中成纤维细胞活化,成为心脏慢性同种异体移植排斥的重要原因[42]。
1.5T细胞的旁分泌信号对心肌纤维化的调控在心肌损伤后,作为对心肌抗原(外源性或内源性)的反应,T细胞的募集可以通过直接的细胞毒性或通过细胞间的信号增强其他类型心脏细胞的活性来引发或促进慢性炎症过程[43]。多项研究表明T细胞浸润广泛存在于由各种缺血性心肌病、非缺血性心肌病(主动脉缩窄、心肌炎等)诱导的心肌纤维化过程中,是心脏发生左心室纤维化和功能障碍的重要条件[44-46]。在此过程中,T细胞与其他细胞间的特异性相互作用可能在ECM重塑中发挥关键作用。
辅助T细胞(helper T cells, Th)是T细胞诸多亚群中的具有协助体液免疫和细胞免疫功能的一大类,因其细胞表面表达CD4又被称为CD4+T细胞[47]。心肌梗死条件下,Th以旁分泌途径释放富含miR-142-3p的Th源外泌体,miR-142-3p进入成纤维细胞后与腺瘤性结肠息肉病蛋白(adenomatous polyposis coli, APC)的转录本结合抑制其表达,进而调控APC/β-catenin信号通路发挥促进成纤维细胞活化的作用[48]。Th又可分为Th1、Th2、Th17和调节T细胞(Treg)等多个Th亚群。既往研究表明,Th17细胞聚集能够促进心肌纤维化,而Treg是心肌纤维化的负向调节细胞[49-50]。因此Th17/Treg动态平衡的打破是心肌纤维化的重要病理因素。研究表明Th17/Treg平衡对心肌纤维化的调控依赖于促纤维化蛋白赖氨酰氧化酶(lysyl oxidase,LOX)的表达,Th17合成的IL-17能够以旁分泌形式与成纤维细胞的IL17受体(IL17 receptor, IL17R)结合,激活细胞外信号调节激酶1/2(extracellular signal-regulated kinase 1/2, ERK1/2)/活化蛋白1(activator protein-1, AP-1)信号通路促进LOX的表达,Treg能够通过旁分泌途径释放大量的IL-10,IL-10与成纤维细胞的IL-10受体(interleukin-10 receptor, IL10R)结合以磷酸化形式激活JAK1/STAT3信号通路抑制LOX的表达[51]。因此,Th17/Treg两种细胞之间的平衡共同通过旁分泌途径对LOX的调控影响了心肌纤维化的进程。此外,有研究向血管紧张素/去甲肾上腺素诱导的心肌纤维化小鼠过继性转移一种工程T细胞—— FAP-CAR T[该T细胞能够表达针对成纤维细胞活化蛋白(FAP)的嵌合受体蛋白(CAR)]显著抑制了心肌纤维化的进展,其具体机制为:FAP仅在活化的心肌成纤维细胞中表达,而FAP-CAR T能够靶向并消除活化的心肌成纤维细胞[52]。这为通过构建工程T细胞靶向抑制或消除活化心肌成纤维细胞从而抑制心肌纤维化提供了思路。
2 基于旁分泌信号调控心肌纤维化的策略
目前基于调控旁分泌信号改善心肌纤维化的研究仍局限于实验阶段,当前已有研究的方法主要包括:抑制或促进旁分泌源细胞释放旁分泌信号以调控心肌纤维化、通过拮抗剂等方式阻断旁分泌信号、利用外源性旁分泌信号实现对心肌纤维化的改善作用等。Hermida等[12]的研究显示心肌细胞β3-肾上腺素能受体(β3-adrenergic receptor, β3AR)与神经元一氧化氮合酶(neuronal nitric oxide synthase, nNOS)的偶联能够显著抑制由ROS诱导的心肌细胞CTGF的上调,从而抑制CTGF通过旁分泌促进心肌成纤维细胞活化的作用。Li等[15]通过遗传修饰、慢病毒携带、miRNA模拟物(miRNA agomir)3种手段分别对大鼠的心肌细胞进行miR-30d的过表达,均改善了心肌梗死引起的心肌纤维化。Yuan[18]等使用miR-378的miRNA agomir显著抑制了成纤维细胞的活化,而在心肌细胞的培养上清中加入miR-378的拮抗剂后(antagomir),心肌细胞通过旁分泌对成纤维细胞活性的抑制作用显著下调。Snail的抑制剂罗格列酮(rosiglitazone)能够通过抑制Snail诱导的旁分泌信号CTGF的上调,改善了心肌梗死大鼠的心脏的纤维化水平[7]。Zhang等[26]给予大鼠ET-1受体拮抗剂(Bosentan)和ET-1中和抗体(ET-1 NAb),结果显示二者均能改善由ET-1高表达诱导的心肌纤维化。另有研究通过表达成纤维细胞中miR-142-3p的靶向蛋白APC,Th来源的miR-142-3p的促成纤维细胞活化能力显著减弱[48]。由此可见,阻断旁分泌信号的方式既包括拮抗旁分泌信号因子也可以通过调控旁分泌信号因子的靶向目标实现。此外,Ramanujam等[53]研究显示:与对照组相比接受主动脉缩窄术的小鼠心肌组织表现出显著的心肌纤维化以及miR-21上调,而存在巨噬细胞基因特异性缺陷(cKO)的小鼠术后的心肌纤维化水平显著低于模型组并且心功能也受到保护,随后将接受脂多糖刺激后的巨噬细胞和静态心肌成纤维细胞进行共培养,结果显示通过锁核酸(LNA)对巨噬细胞的沉默能够显著抑制其通过旁分泌信号诱导的心肌成纤维细胞活化。表明在心肌纤维化过程中,巨噬细胞通过旁分泌促心肌成纤维细胞活化的作用可能具有某些miRNA依赖性,这也成为了心肌纤维化潜在的治疗靶点。
3 总结
心肌纤维化是心脏老化或损伤后的基础病理表现,也是影响心功能的重要因素。心脏中非成纤维细胞通过旁分泌途径与成纤维细胞以及其他非成纤维细胞间的信号传递参与了对心肌纤维化的调控。深入刨析非成纤维细胞通过旁分泌信号构建的心肌纤维化调控网络,能够扩展心肌纤维化潜在的治疗靶点。如本综述所讨论,外界刺激下内皮细胞除通过分泌CTGF、ET-1促进成纤维细胞的活化外,其还能通过促进心肌细胞的凋亡以及内皮细胞自身向成纤维细胞转化等多途径推动心肌纤维化的进展。中性粒细胞和Th17细胞也能分别通过分泌MPO和IL17激活成纤维细胞。而嗜酸性粒细胞和Treg细胞分别通过分泌ECP和IL10能够抑制成纤维细胞的活化。巨噬细胞既能够分泌TGF-β1促进内皮细胞发生EndMT,也能够分泌circUbe3a促进成纤维细胞向肌成纤维细胞转化参与心肌纤维化的进程。值得注意的是,心脏非成纤维细胞的旁分泌效应对心肌纤维化的调控可能具有双向作用。例如心肌细胞既能够通过分泌CTGF激活成纤维细胞,也能够分泌CT或miRNA(miR-30d和miR-378等)抑制成纤维细胞的活化。这体现了细胞间信号传递对于心肌纤维化的调控本身具有一个复杂的拮抗机制,而两种对立效应之间的平衡状态决定了心肌纤维化的最终进展(图1)。
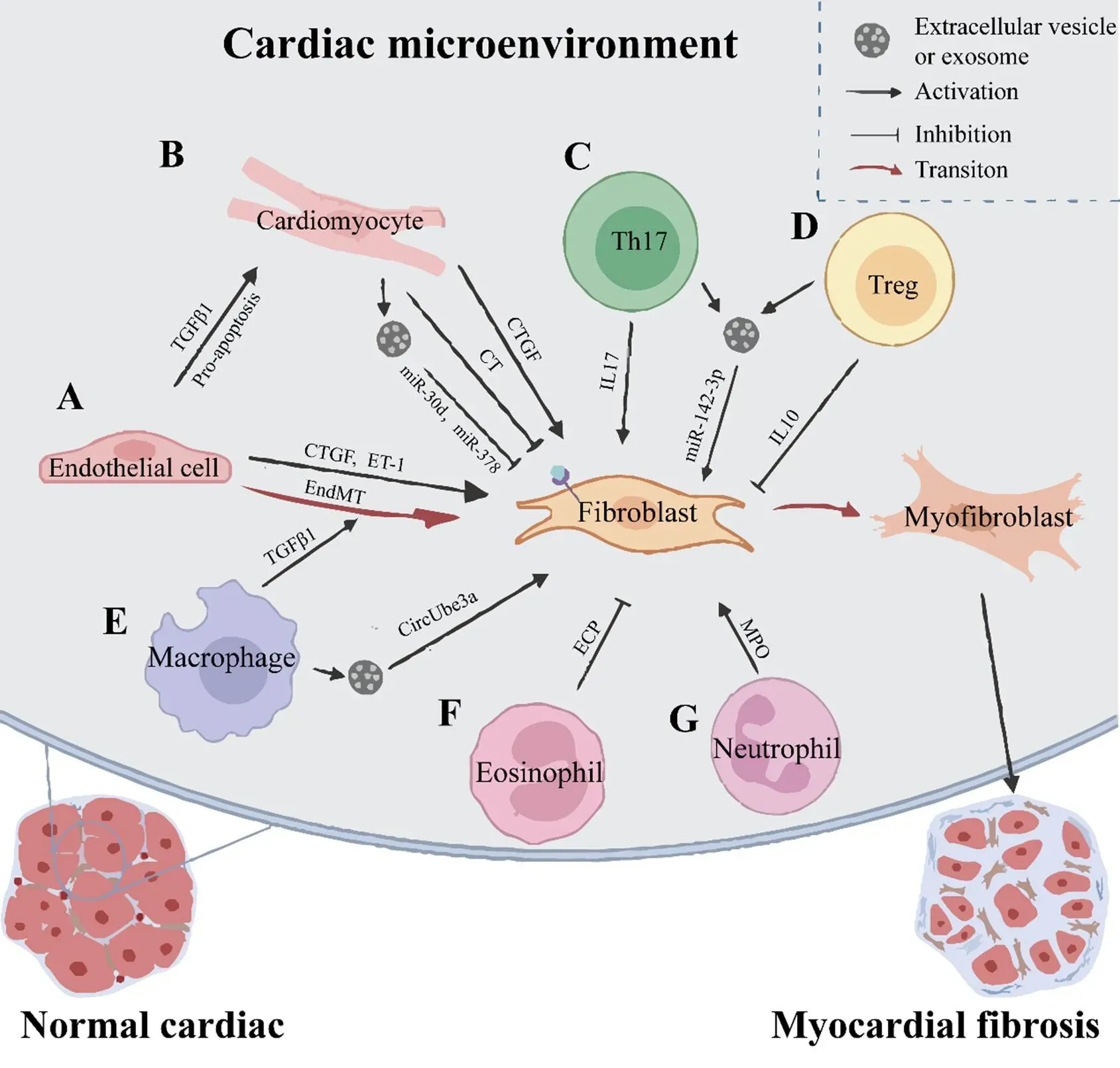
Figure 1. Paracrine signaling in cardiac microenvironment regulated myocardial fibrosis. A: endothelial cell secreted connective tissue growth factor (CTGF)[7] and endothelin-1 (ET-1)[26] to promote the activation of fibroblasts, and secreted transforming growth factor β1 (TGFβ1)[27] to promote the apoptosis of cardiomyocytes; B: cardiomyocyte secreted CTGF[12] to promote the activation of fibroblasts, and secreted miR-30d[15], miR-378[18] and calcitonin (CT)[21] to inhibit the activation of fibroblasts; C and D: T helper cell (Th) secreted miR-142-3p[48] to promote the activation of fibroblasts, and interleukin-17 (IL17) secreted by T helper 17 cell (Th17) and IL10 secreted by regulatory T cell (Treg) co-regulated fibroblast activity[51]; E: macrophage secreted circUbe3a[34] to promote fibroblast activity, and secreted TGFβ1 to promote the process of endothelial-mesenchymal transition (EndMT)[31]; F: eosinophil secreted eosinophil cationic protein (ECP)[40] to inhibit the activation of fibroblasts; G: neutrophil secreted myeloperoxidase (MPO)[36] to promote fibroblast activity.
综上所述,非成纤维细胞通过旁分泌途径释放大量的信号载体(蛋白、miRNA、circRNA等)与微环境中的其他细胞形成密集的通讯网络,细胞间相互影响,共同实现对心肌纤维化的调控。然而,基于调控旁分泌信号网络以实现改善心肌纤维化的策略仍处于实验阶段,因此需要进一步的研究并开发能够应用于临床的旁分泌调控制剂,以达到改善和治疗心肌纤维化的目的。
[1] Bozkurt B, Coats AJS, Tsutsui H, et al. Universal definition and classification of heart failure: a report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure: Endorsed by the Canadian Heart Failure Society, Heart Failure Association of India, Cardiac Society of Australia and New Zealand, and Chinese Heart Failure Association[J]. Eur J Heart Fail, 2021, 23(3):352-380.
[2] Frangogiannis NG. Cardiac fibrosis[J]. Cardiovasc Res, 2021, 117(6):1450-1488.
[3] Gourdie RG, Dimmeler S, Kohl P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease[J]. Nat Rev Drug Discov, 2016, 15(9):620-638.
[4] Nagaraju CK, Robinson EL, Abdesselem M, et al. Myofibroblast phenotype and reversibility of fibrosis in patients with end-stage heart failure[J]. J Am Coll Cardiol, 2019, 73(18):2267-2282.
[5] Stratton MS, Bagchi RA, Felisbino MB, et al. Dynamic chromatin targeting of BRD4 stimulates cardiac fibroblast activation[J]. Circ Res, 2019, 125(7):662-677.
[6] Lafuse WP, Wozniak DJ, Rajaram MVS. Role of cardiac macrophages on cardiac inflammation, fibrosis and tissue repair[J]. Cells, 2020, 10(1):51.
[7] Lee SW, Won JY, Kim WJ, et al. Snail as a potential target molecule in cardiac fibrosis: paracrine action of endothelial cells on fibroblasts through snail and CTGF axis[J]. Mol Ther, 2013, 21(9):1767-1777.
[8] Keshtkar S, Azarpira N, Ghahremani MH. Mesenchymal stem cell-derived extracellular vesicles: novel frontiers in regenerative medicine[J]. Stem Cell Res Ther, 2018, 9(1):63.
[9] Prabhu SD, Frangogiannis NG. The Biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis[J]. Circ Res, 2016, 119(1):91-112.
[10] Frangogiannis NG. Regulation of the inflammatory response in cardiac repair[J]. Circ Res, 2012, 110(1):159-173.
[11] Panizzi P, Swirski FK, Figueiredo JL, et al. Impaired infarct healing in atherosclerotic mice with Ly-6Chimonocytosis[J]. J Am Coll Cardiol, 2010, 55(15):1629-1638.
[12] Hermida N, Michel L, Esfahani H, et al. Cardiac myocyte β3-adrenergic receptors prevent myocardial fibrosis by modulating oxidant stress-dependent paracrine signaling[J]. Eur Heart J, 2018, 39(10):888-898.
[13] Xue R, Tan W, Wu Y, et al. Role of exosomal miRNAs in heart failure[J]. Front Cardiovasc Med, 2020, 7:592412.
[14] Lu TX, Rothenberg ME. MicroRNA[J]. J Allergy Clin Immunol, 2018, 141(4):1202-1207.
[15] Li J, Salvador AM, Li G, et al. Mir-30d regulates cardiac remodeling by intracellular and paracrine signaling[J]. Circ Res, 2021, 128(1):e1-e23.
[16] Song S, Liu L, Yu Y, et al. Inhibition of BRD4 attenuates transverse aortic constriction- and TGF-β-induced endothelial-mesenchymal transition and cardiac fibrosis[J]. J Mol Cell Cardiol, 2019, 127:83-96.
[17] Yao Y, Hu C, Song Q, et al. ADAMTS16 activates latent TGF-β, accentuating fibrosis and dysfunction of the pressure-overloaded heart[J]. Cardiovasc Res, 2020, 116(5):956-969.
[18] Yuan J, Liu H, Gao W, et al. MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress[J]. Theranostics, 2018, 8(9):2565-2582.
[19] Reese-Petersen AL, Olesen MS, Karsdal MA, et al. Atrial fibrillation and cardiac fibrosis: a review on the potential of extracellular matrix proteins as biomarkers[J]. Matrix Biol, 2020, 91/92:188-203.
[20] Maleitzke T, Hildebrandt A, Dietrich T, et al. The calcitonin receptor protects against bone loss and excessive inflammation in collagen antibody-induced arthritis[J]. iScience, 2022, 25(1):103689.
[21] Moreira LM, Takawale A, Hulsurkar M, et al. Paracrine signalling by cardiac calcitonin controls atrial fibrogenesis and arrhythmia[J]. Nature, 2020, 587(7834):460-465.
[22] Wang Y, Zhang J, Wang Z, et al. Endothelial-cell-mediated mechanism of coronary microvascular dysfunction leading to heart failure with preserved ejection fraction[J/OL]. Heart Fail Rev, 2022 (2022-03-09) [2022-07-14]. https://link.springer.com/article/10.1007/s10741-022-10224-y.
[23] Liu ZH, Zhang Y, Wang X, et al. SIRT1 activation attenuates cardiac fibrosis by endothelial-to-mesenchymal transition[J]. Biomed Pharmacother, 2019, 118:109227.
[24] Wilhelmi T, Xu X, Tan X, et al. Serelaxin alleviates cardiac fibrosis through inhibiting endothelial-to-mesenchymal transition via RXFP1[J]. Theranostics, 2020, 10(9):3905-3924.
[25] 汤石林, 刘一剑, 彭良善, 等. 大鼠急性心肌梗死后NLRP3炎性体-IL-1β信号轴激活与End-MT的关系[J]. 中国病理生理杂志, 2018, 34(11):1935-1939.
Tang S, Liu Y, Peng L, et al. Relationship between NLRP3 inflammasome-IL-1β axis and End-MT after acute myocardial infarction in rats[J]. Chin J Pathophysiol, 2018, 34(11):1935-1939.
[26] Zhang X, Hu C, Yuan YP, et al. Endothelial ERG alleviates cardiac fibrosis via blocking endothelin-1-dependent paracrine mechanism[J]. Cell Biol Toxicol, 2021, 37(6):873-890.
[27] Sniegon I, Prieß M, Heger J, et al. Endothelial mesenchymal transition in hypoxic microvascular endothelial cells and paracrine induction of cardiomyocyte apoptosis are mediated via TGFβ₁/SMAD signaling[J]. Int J Mol Sci, 2017, 18(11):2290.
[28] Dick SA, Macklin JA, Nejat S, et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction[J]. Nat Immunol, 2019, 20(1):29-39.
[29] 覃宇燕, 吴茜, 余细勇. 单核/巨噬细胞调控心梗后心肌纤维化的研究进展[J]. 中国病理生理杂志, 2020, 36(8):1521-1525.
Qin YY, Wu Q, Yu XY. Progress in monocytes/macrophages regulating cardiac fibrosis after myocardial infarction[J]. Chin J Pathophysiol, 2020, 36(8):1521-1525.
[30] O'Rourke SA, Dunne A, Monaghan MG. The role of macrophages in the infarcted myocardium: orchestrators of ECM remodeling[J]. Front Cardiovasc Med, 2019, 6:101.
[31] Alonso-Herranz L, Sahún-Español Á, Paredes A, et al. Macrophages promote endothelial-to-mesenchymal transition via MT1-MMP/TGFβ1 after myocardial infarction[J]. Elife, 2020, 9:e57920.
[32] Cheng R, Dang R, Zhou Y, et al. MicroRNA-98 inhibits TGF-β1-induced differentiation and collagen production of cardiac fibroblasts by targeting TGFBR1[J]. Hum Cell, 2017, 30(3):192-200.
[33] Patop IL, Wüst S, Kadener S. Past, present, and future of circRNAs[J]. EMBO J, 2019, 38(16):e100836.
[34] Wang Y, Li C, Zhao R, et al. CircUbe3a from M2 macrophage-derived small extracellular vesicles mediates myocardial fibrosis after acute myocardial infarction[J]. Theranostics, 2021, 11(13):6315-6333.
[35] Okyere AD, Tilley DG. Leukocyte-dependent regulation of cardiac fibrosis[J]. Front Physiol, 2020, 11:301.
[36] Mollenhauer M, Bokredenghel S, Geißen S, et al. Stamp2 protects from maladaptive structural remodeling and systolic dysfunction in post-ischemic hearts by attenuating neutrophil activation[J]. Front Immunol, 2021, 12:701721.
[37] Mollenhauer M, Friedrichs K, Lange M, et al. Myeloperoxidase mediates postischemic arrhythmogenic ventricular remodeling[J]. Circ Res, 2017, 121(1):56-70.
[38] Baandrup U. Eosinophilic myocarditis[J]. Herz, 2012, 37(8):849-852.
[39] Diny NL, Baldeviano GC, Talor MV, et al. Eosinophil-derived IL-4 drives progression of myocarditis to inflammatory dilated cardiomyopathy[J]. J Exp Med, 2017, 214(4):943-957.
[40] Liu J, Yang C, Liu T, et al. Eosinophils improve cardiac function after myocardial infarction[J]. Nat Commun, 2020, 11(1):6396.
[41] Yang C, Li J, Deng Z, et al. Eosinophils protect pressure overload- and β-adrenoreceptor agonist-induced cardiac hypertrophy[J/OL]. Cardiovasc Res, 2022 (2022-04-08) [2022-06-21]. https://academic.oup.com/cardiovascres/advance-article/doi/10.1093/cvr/cvac060/6565299.
[42] Schiechl G, Hermann FJ, Rodriguez Gomez M, et al. Basophils trigger fibroblast activation in cardiac allograft fibrosis development[J]. Am J Transplant, 2016, 16(9):2574-2588.
[43] Blanton RM, Carrillo-Salinas FJ, Alcaide P. T-cell recruitment to the heart: friendly guests or unwelcome visitors?[J]. Am J Physiol Heart Circ Physiol, 2019, 317(1):H124-H140.
[44] Myers JM, Cooper LT, Kem DC, et al. Cardiac myosin-Th17 responses promote heart failure in human myocarditis[J]. JCI Insight, 2016, 1(9):e85851.
[45] Saxena A, Dobaczewski M, Rai V, et al. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function[J]. Am J Physiol Heart Circ Physiol, 2014, 307(8):H1233-H1242.
[46] Nevers T, Salvador AM, Grodecki-Pena A, et al. Left ventricular T-Cell recruitment contributes to the pathogenesis of heart failure[J]. Circ Heart Fail, 2015, 8(4):776-787.
[47] Hirahara K, Nakayama T. CD4+T-cell subsets in inflammatory diseases: beyond the Th1/Th2 paradigm[J]. Int Immunol, 2016, 28(4):163-171.
[48] Cai L, Chao G, Li W, et al. Activated CD4+T cells-derived exosomal miR-142-3p boosts post-ischemic ventricular remodeling by activating myofibroblast[J]. Aging (Albany NY), 2020, 12(8):7380-7396.
[49] Chang SL, Hsiao YW, Tsai YN, et al. Interleukin-17 enhances cardiac ventricular remodeling via activating MAPK pathway in ischemic heart failure[J]. J Mol Cell Cardiol, 2018, 122:69-79.
[50] Liu Y, Lu H, Zhang C, et al. Recent advances in understanding the roles of T cells in pressure overload-induced cardiac hypertrophy and remodeling[J]. J Mol Cell Cardiol, 2019, 129:293-302.
[51] Lu M, Qin X, Yao J, et al. Th17/Treg imbalance modulates rat myocardial fibrosis and heart failure by regulating LOX expression[J]. Acta Physiol (Oxf), 2020, 230(3):e13537.
[52] Aghajanian H, Kimura T, Rurik JG, et al. Targeting cardiac fibrosis with engineered T cells[J]. Nature, 2019, 573(7774):430-433.
[53] Ramanujam D, Schön AP, Beck C, et al. MicroRNA-21-dependent macrophage-to-fibroblast signaling determines the cardiac response to pressure overload[J]. Circulation, 2021, 143(15):1513-1525.
Progress in regulation of myocardial fibrosis by paracrine signaling of cardiac non-fibroblasts
ZHOU Zhou1, ZHANG Ke-ke1, WANG Zhen2, WANG Yong2, LIU Yang2△
(1,,250000,;2,,250000,)
Myocardial fibrosis is the result of extracellular matrix (ECM) remodeling after aging or myocardial injury, and it is also the main pathological feature accompanying cardiac dysfunction. Cardiac fibroblasts, responsible for the synthesis and accumulation of collagen fibers, are the central cell group for myocardial fibrosis. However, in recent years, it has been found that the signal communication between cardiac non-fibroblasts (including myocardial cells, myocardial microvascular endothelial cells, inflammatory cells and so on) and cardiac fibroblasts as well as between cardiac non-fibroblasts through the paracrine pathway plays an important role in the derivation and activation of cardiac fibroblasts, the change of ECM and the progression of cardiac fibrosis. This review focuses on the effects of paracrine signaling of cardiac non-fibroblasts on cardiac cell populations, so as to unveil the communication network of cardiac microenvironment during myocardial fibrosis and provide more references for the study of pathological mechanism of myocardial fibrosis.
Myocardial fibrosis; Cardiac non-fibroblasts; Paracrine
1000-4718(2022)09-1709-07
2022-05-09
2022-07-26
18678850885; E-mail: 18678850885@163.com
R542.2+3; R363
A
10.3969/j.issn.1000-4718.2022.09.022
[基金项目]国家自然科学基金资助项目(No. 82104797);山东省自然科学基金(No. ZR2020MR333; No. ZR2021LZY011);山东省中医药科技发展计划(No. 2015-088)
(责任编辑:宋延君,罗森)
