西藏藏族Cornelia de Lange综合征1例病例报告并文献复习
2022-09-26田亚平张春燕
达 珍,马 雅,孟 岩,田亚平,张春燕
1 西藏自治区妇产儿童医院 儿科,西藏拉萨 850015;2 西藏自治区第二人民医院 儿科,西藏拉萨 850000;3 解放军总医院第一医学中心 儿科,北京 100853;4 解放军总医院医学创新研究部 出生缺陷防治技术研究中心,北京 100853
Cornelia de Lange综 合 征(Cornelia de Lange syndrome,CdLS;OMIM #122470,#300590,#610759)是一种罕见的累及多器官系统的遗传性疾病,1933年由荷兰儿科医生Cornelia de Lange首次系统报道,之后以他的名字命名此病。目前其发病率各国报道不一致(1/10 000 ~ 1/50 000),常呈散发,男女发病率基本相同。已发现CdLS致病基因有NIPBL、SMC1A、SMC3、RAD21、HDAC8等[1-2],常染色体显性遗传或X连锁遗传。CdLS有典型和轻型两种临床表型,典型CdLS的表现有特殊面容、四肢异常、神经发育迟滞。面部特征包括连眉、浓眉、睫毛长而弯曲,短鼻、鼻孔朝前等。神经发育异常表现为智力低下、行动呆板,部分病例伴自闭症、自残行为和癫痫发作。胎儿宫内及出生后发育迟滞。其他器官发育异常还包括先天性膈疝、先天性心脏病、腭裂、生殖器异常、泌尿道畸形、胃肠道异常、大理石样皮肤、多毛、脊柱侧弯、脑脊膜膨出等[3-5]。死亡原因在新生儿期多为宫内发育迟滞、早产、复杂的先天性心脏病,在婴幼儿期常因胃肠道异常引起反流和吸入性肺炎导致死亡。本文报道1例在西藏自治区拉萨市附近发现的典型婴儿CdLS病例,虽然国内已有报道110例[6-7],但本例是迄今发现并报道的第1例藏族病例。除具典型临床表现外,该例还伴脑部发育异常、隐形脊柱裂等报道较少的中枢神经系统异常表现,基因检测为NIPB基因杂合突变。
1病例资料患儿女性,出生55 d,藏族,因“呕吐、消瘦55 d”于西藏自治区第二人民医院儿科(2018年)就诊;孕34周早产,出生体质量2 030 g。出生时无产伤,Apgar评分1 min 8分,3 min 9分,5 min 10分,母乳喂养,母亲无妊娠相关疾病。患儿母亲27岁,G2P2,第一胎男孩,孕39周足月出生,出生体质量5 250 g,有严重唇腭裂(已行修补术)现无异常。第一、二胎均未进行产前检查。其父30岁,体健,无烟酒嗜好。父母均为当地农民,双亲均无农药、毒药、重金属等特殊毒物接触史,否认近亲结婚。奶奶为耳聋患者。其他无特殊家族史。
体格检查:体温36.6℃,呼吸30次/min,心率120/min,血氧饱和度90%,体质量2 050 g,头围32 cm,胸围30 cm,身长46 cm(均低于同龄儿正常值的第3百分位以下)。特殊外貌:连眉,眼球突出,瞳孔小,头发浓密,前囟2 cm × 2 cm,睫毛长而卷曲,塌鼻梁,鼻孔前倾,人中长,上唇薄而下翻呈鱼嘴样,高颧弓,小颌,下颌耻合处突起,耳低位,颈短(图1A)。皮肤多毛,大理石样纹理(图1B)。乳头和脐发育不良,双肺呼吸音清,未闻及干湿性啰音。心前区无隆起,各瓣膜听诊区未闻及病理性杂音,腹部无异常。上肢短,双肘屈曲(图1C)拇指近位,第五指侧弯,指甲薄而小(图1E),足细小。四肢肌张力增高,脊柱无侧弯(图1F),骶尾处可见隐形脊柱裂(图1D),哭声调低沙哑,呕吐频繁。
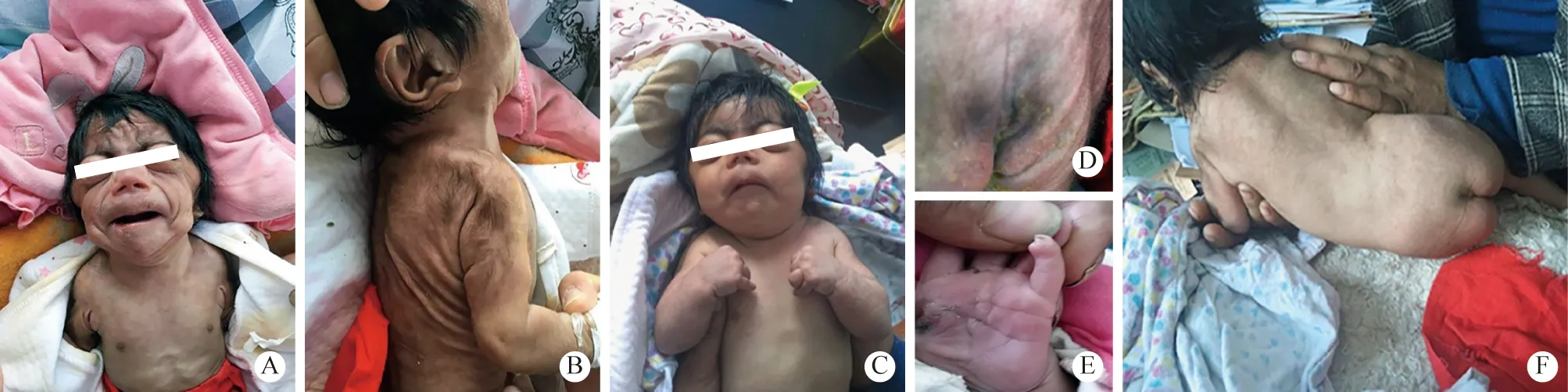
图1 患儿的临床表现 A:患儿有特殊面容,眉毛拱起,眼球突出,头发浓密,鼻孔前倾,鼻子短而宽,鼻梁凹陷,人中长而突出,嘴唇薄,嘴角向下(鲤鱼嘴),颧弓高,小颌畸形,下巴肿胀,短颈;B:耳位低,多毛,大理石样纹路;C:前臂短,双肘屈折;D:骶尾部隐性脊柱裂;E:拇指近端和五指侧弯,指甲薄而小;F:无脊柱侧凸
辅助检查:超声心动图未见异常,腹部超声未见双肾积水,肾功能、电解质正常,血常规正常。胸部X线片未见异常,上肢X线片提示左侧肘关节脱位(图2)。血生化检查,谷丙转氨酶升高,75 IU/L(参考值为5 ~ 45 IU/L),谷草转氨酶升高,72 IU/L(参考值为8 ~ 40 IU/L);肌酸激酶升高,250 U/L(参考值为26 ~ 174 U/L),肌酸激酶同工酶36 U/L(参考值为2 ~ 24 U/L);免疫球蛋白G 7.4 mg/dL(参考值为8 ~ 16 mg/dL), 免疫球蛋白A 0.00 mg/dL(参考值为0.7 ~ 3.3 mg/dL)。脊柱及头部MRI:脑白质发育不良(图3);脊柱骶尾部隐形脊柱裂(图1D)。
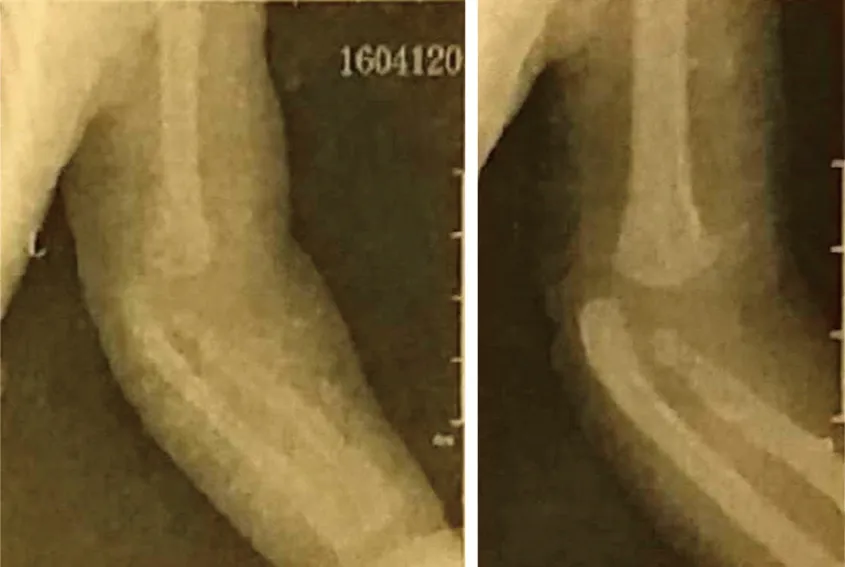
图2 左肘关节脱位X线片(正、侧位)
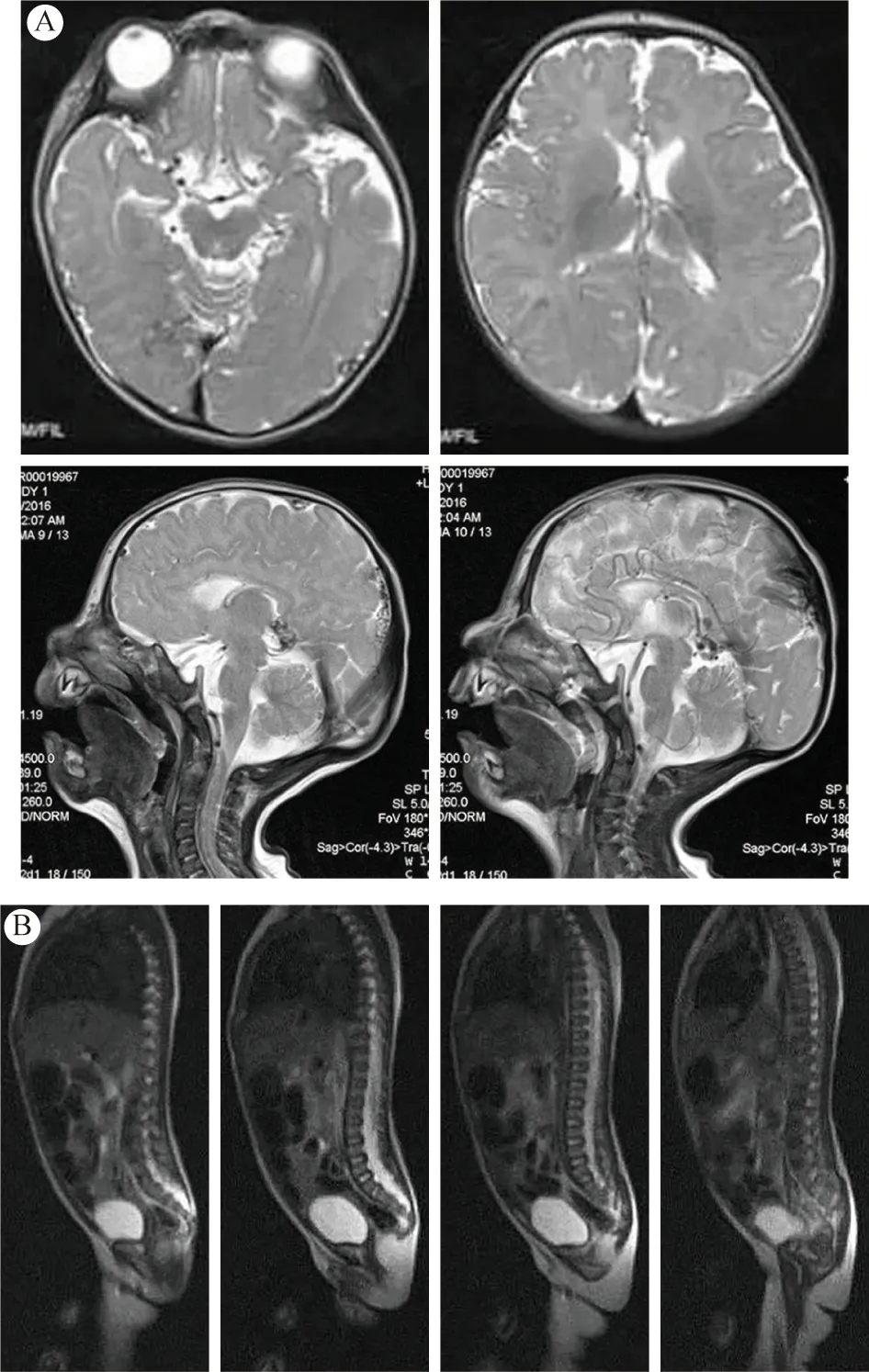
图3 MRI影像结果 A:头部MRI显示脑白质发育不良;B:脊柱MRI显示隐形脊柱裂
诊断:当地医院向解放军总医院申请会诊,经解放军总医院第一医学中心儿科和神经内科专家会诊,在出生缺陷防控技术研究中心行48种新生儿代谢疾病筛查和基因诊断。代谢疾病筛查结果正常,基因检测(家系)结果显示NIPBL基因杂合突变(图4)。依据Kline等[8]以及美国CdLS基金会和世界CdLS组织报道的诊断标准和评分系统,诊断患儿为CdLS。
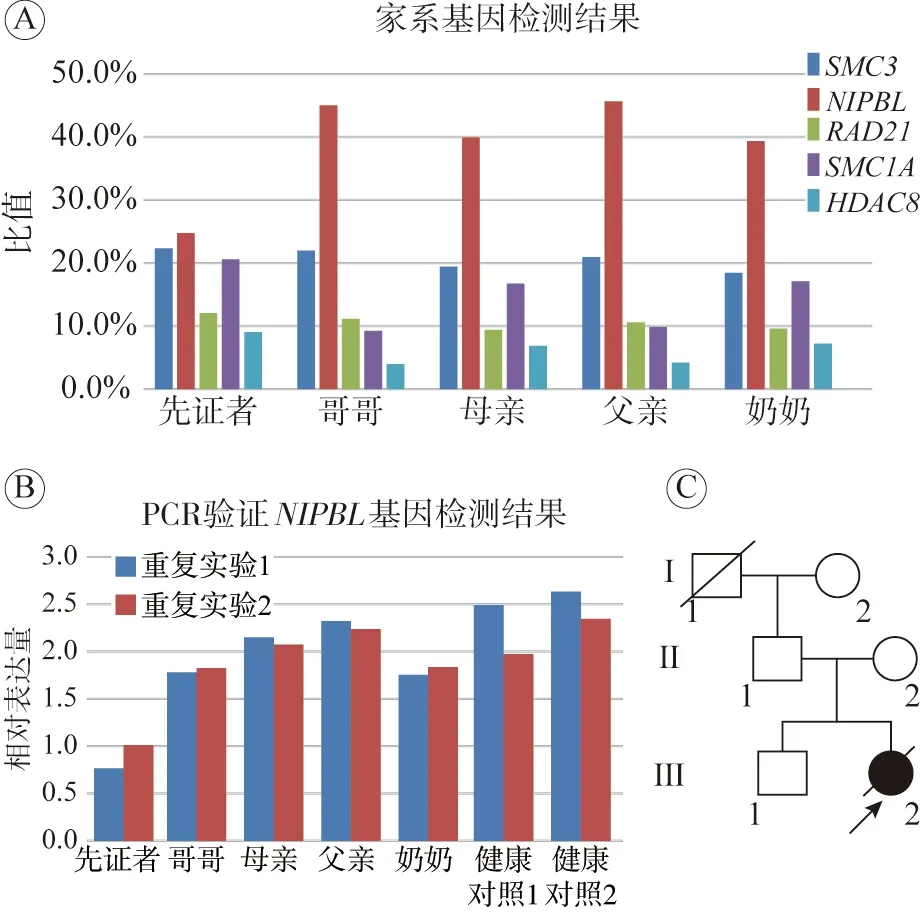
图4 基因检测结果A:患儿家系CdLS相关基因检测结果,患儿(先证者)NIPBL基因表达量与其他家系成员相比减少一半,为阳性,其他家系成员均为阴性;B:患儿家系NIPBL基因检测结果;C:家系分析图
治疗:入院后给予留置胃管喂养,静脉给予能量合剂、小儿氨基酸、脂肪乳、水乐维他等支持治疗,呕吐减轻,体质量增至2 200 g,身长46 cm,经治疗14 d后好转出院。
随访:患儿出院后仍然间断吐奶,但呕吐量和次数均较前明显减少,哭声仍沙哑调低,2月龄、3月龄、4月龄时随访,体质量分别为2 300 g、2 350 g、2 700 g,身长分别为46.5 cm、47 cm、47.5 cm。其母用当地手法(菜籽油)每天进行抚触,3个月时体毛大量脱落。患儿8月龄时死亡,具体死亡原因不详。
2 讨论截至2022年4月,在中国知网和Pubmed数据库检索相关文献,检索到47篇 110例国内CdLS病例报道,其中3篇为英文文献[9]。1982年报道了国内首例CdLS[10],之后涉及眼科、耳鼻喉、皮肤相关报道4篇。全部患儿均因有典型的临床表现而被诊断。因各种原因,已报道的病例基因检测率和检出率较低,110例中56例行基因检 测,NIPBL突 变 占78.57%(44/56),SMC1A突变占7.14%(4/56),HDAC8突变占5.36%(3/56),RAD21突变占1.79%(1/56)[11-14]。全部患儿后期随访率差,远期进展情况不详。
CdLS报道迄今已有100余年,据统计国外发现约有500多例患者[6-7]。该综合征典型和轻型均具有特征性面部表现。典型CdLS还有四肢、心脏异常以及较严重的生长发育和神经精神发育迟滞。轻型CdLS很少有四肢、心脏异常,神经精神缺陷相对不明显。本例为藏族,临床表现大部分符合典型特点,突出的有典型的面部特征,多毛,上肢短小屈曲,第五指侧弯,肌张力高,皮肤大理石样花纹,骶尾部隐形脊柱裂,但无明显心脏、肾、生殖器异常。近年来对CdLS的神经精神系统评价越来越受重视。Silva-Hernández等[15]报道中枢神经系统影像学用于CdLS评价神经系统发育障碍;Mahdi和Whitehead[16]报道大脑和脊髓在影像学中的多种异常,Roshan Lal等[17]报道67%的患者脑部MRI存在异常情况,包括脑萎缩、脑白质改变、小脑发育不全、脑室扩大等。国内文献也有脑部异常的报道,本例脑部及脊柱MRI均存在异常,经解放军总医院神经外科专家会诊,患儿脑白质、脊髓腰骶发育异常,骶尾部存在隐形脊柱裂,符合典型病例特点。消化系统异常在本例较突出,表现从出生至今持续呕吐,但辅助检查未能全面进行,所以无异常报道,文献报道通常有胃-食管反流、肠套叠、肠扭转、幽门狭窄、肛门闭锁、先天膈疝等,误吸和吸入性肺炎常导致患儿死亡。患儿55 d时血氧饱和度90%,与当地健康同龄儿相似,考虑与拉萨地区海拔较高相关。该综合征存在多个系统的畸形和异常,随着对疾病的不断认识,一些罕见的临床表现可能会不断的被发现报道,同时这也将为指导治疗和注意事项提供依据。
研究表明,该病病因与基因突变有关,故基因检测至关重要,到目前已发现有常染色体显性(NIPBL、SMC3、RAD21)或X连锁(SMC1A和HDAC8)的5种致病基因。发病机制研究发现它们均参与构成或调节Cohesin复合体,故与Cohesin复合体构成相关的基因突变都有可能与CdLS发病相关,约占70%的病例。Mannini[18]报道目前为止已发现311种突变,包括错义突变、无义突变、缺失、插入、剪切位点突变和基因重组。其中NIPBL和HDAC8突变与典型病例相关,SMC1A、SMC3、RAD21与轻型病例相关。NIPBL编码的蛋白Delangin属于染色体黏附家族,在成年人和胎儿的各个组织均有表达,尤其是上肢、腮弓和颅面间质,参与骨骼和软组织(面部、下颌、四肢)的构成[19]。国内已报道的病例NIPBL基因突变占78.57%(44/56)。本例检测为NIPBL基因突变,突变位置chr5: 36876861-37065926,变异类型为杂合突变,变异状态为致病突变,其父母、哥哥和奶奶该基因检测均正常。结合本病例的临床表型符合典型CdLS表型,NIPBL基因杂合突变致病(常染色体显性遗传),考虑患儿NIPBL基因新发突变致病,父母再生育患儿的概率很小。但2018年本病例发现时实验室基因检测手段有限,未行基因测序,后患儿死亡未留存样本,因此不能准确到基因SNP位点,不排除其他未测基因的突变,同时也不能排除患儿父母NIPBL生殖细胞嵌合的可能。
CdLS综合征全世界均有报道,未发现地域和种族差异。目前无特殊治疗方法,所有患者均有消化道异常,需注意因误吸或吸入性肺炎引起死亡。
