SCN1A相关发育性癫痫-运动障碍性脑病1例报告
2022-09-20田茂强赵静杨冰竹雷文婷陈静李娟郎长会束晓梅
田茂强,赵静,杨冰竹,雷文婷,陈静,李娟,郎长会,束晓梅
离子通道与多种细胞的电活动相关,导致癫痫及运动障碍的基因多数与离子通道相关[1]。编码Na+通道α1亚基的SCN1A基因(OMIM* 182389)主要在中枢神经系统表达,参与神经递质释放及调节神经细胞兴奋性,是较早发现的与癫痫相关的基因之一,该基因突变常导致Dravet综合征,使用Na+通道阻滞剂治疗可使该综合征患者癫痫发作加重[2]。有研究显示SCN1A突变还可导致非Dravet综合征癫痫,甚至非痫性发作,但缺乏特征性基因型-表型关系,导致临床诊断困难[3]。本文报道1例SCN1A基因突变,表现为癫痫及非痫性发作、经Na+通道阻滞剂治疗部分有效的患者,总结其临床症状、遗传学及脑电图(EEG)特征,以拓宽临床医生对SCN1A突变基因型-表型关系的认识。
1 病例报告
患儿,男,9月余,因“反复发作性症状9个月”就诊。患儿共有3种发作类型,第一种发作类型在出生3 d即出现,主要表现为睡眠中突然肢体屈曲,双上肢上抬,呼吸暂停,伴或不伴口周发绀,持续1~2 min缓解,缓解后剧烈哭闹,7~10次/d。同期EEG未见明显发作期图形(20日龄)。考虑诊断:(1)癫痫可疑;(2)阵发性运动障碍可疑。试用维生素B6(50 mg/d)治疗5 d无效,加用丙戊酸钠口服液25 mg/(kg·d)治疗1月余,患儿发作性症状无好转。复查EEG(54日龄)捕获到2次发作,发作期EEG表现为症状前2~4 s出现中线区(Fz、Cz、Pz)低波幅9 Hz~10 Hz快波节律(图1,A-B),考虑为癫痫性发作。排除其他病因后确诊为癫痫,故加用奥卡西平联合抗惊厥治疗,加量至27 mg/(kg·d)时发作减少至2~3次/d,最长可3 d无发作。
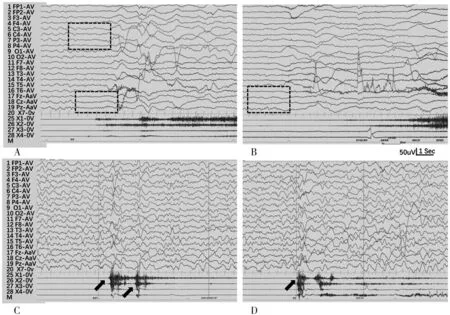
注:A~B为第1种发作类型发作期EEG[症状前数秒先出现低波幅快波节律(虚线框标示),发作期干扰太大无法判断],C~D为第2种发作类型发作期EEG[监测到肢体抖动两下两次(箭头所示),均为非痫性发作]。图1 患儿不同时期发作期EEG
个人史G2P2,足月剖腹产,患儿出生时体质量4 900 g,无窒息抢救史。家族史阴性。
辅助检查:血常规、血糖、肝肾功能、电解质及血乳酸正常,血尿代谢筛查无异常;心脏彩超(-)、心电图正常。头颅磁共振 (4月龄)正常。
结合患儿早发癫痫,无结构、急性炎症及代谢紊乱病因,因此考虑癫痫病因为先天性可能性大,经遵义医科大学附属医院伦理委员会批准,患儿监护人签署知情同意书,采集患儿及父母外周血进行基因全外显子测序。结果显示(3月龄)SCN1A杂合突变(NM_001165963.4:c.485C>T(p.Thr162Ile),父母该位点为野生型(图2),为新生突变。该变异在ClinVar数据库中有1次疑似致病性收录[4],在ExAC、gnomAD、千人基因组亚洲人群数据库中没有收录,3种变异预测软件(SIFT、Polyphen2及MutationTaster)提示该变异为有害性突变。经ACMG(美国医学遗传学与基因组学学会)评级,该突变被评为疑似致病性突变(PS2+PM2+PP2+PP3)。
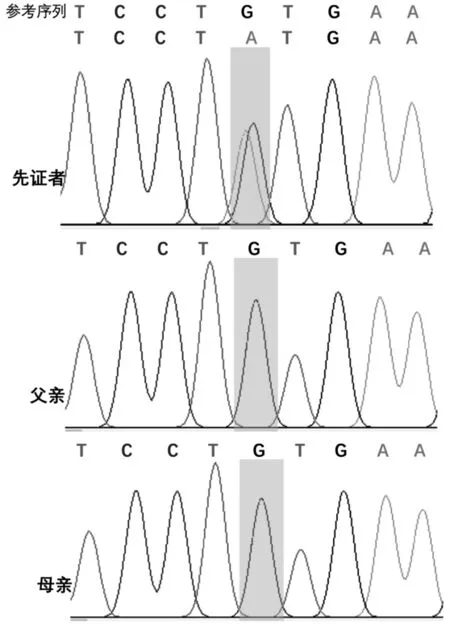
图2 患儿本家系SCN1A一代测序峰图
发现SCN1A致病性突变后停用奥卡西平,患儿发作加重,每日7~8次。同时出现新的发作类型:第2种发作类型表现为多在睡眠中出现的惊跳样动作,单下或连续数下,复查EEG证实为非痫性发作(图1,C-D);第3种发作类型为过度运动:突然双侧肢体“使劲”样动作或伴下肢蹬踏,伴呻吟和(或)哭闹,可持续约40~50 min左右,不伴面色青紫,此种发作共2~3次,EEG未捕获到此种发作。考虑诊断为SCN1A谱系病:(1)癫痫,局灶性发作继发全面强直阵挛发作;(2)阵发性运动障碍。治疗上予奥卡西平小剂量加回[20 mg/(kg·d)],发作明显减少。因癫痫未控制及较多的运动障碍发作,故就诊于外院,予减停丙戊酸钠及奥卡西平,加用左乙拉西坦[44 mg/(kg·d)]抗惊厥治疗,但第2种发作类型仍较频繁,第3种发作类型偶发,故渐加用托吡酯及氯巴占联合抗癫痫治疗。随访至9月龄(2022年3月1日),近2个月未观察到第1种发作类型,第2种发作类型每2~3 d发作1次。发育落后明显,以运动落后显著,抬头可,仍不能独坐,能逗笑,追声追物可。体格检查:头围44 cm,体质量9.5 kg,抬头可,无特殊面容,皮肤无色素脱失斑、牛奶咖啡斑,心肺腹无异常,四肢肌张力稍高,病理征阴性。目前用药剂量为左乙拉西坦5 mL/d[52.6 mg/(kg·d)]、托吡酯75 mg/d[7.8 mg/(kg·d)]及氯巴占7.5 mg/d[0.78 mg/(kg·d)]。
2 讨论
SCN1A定位于2q24.3,编码神经元电压门控Na+通道 Nav1.1,具有4个同源结构域(DI-DIV),每个结构域包含6个跨膜区(S1-S6),其中S4为电压感受区,该区域的突变通常导致功能缺失(loss of function,LOF)而产生较重的癫痫表型如Dravet综合征[2,5-6]。在动物模型研究中也证实SCN1A通过LOF性突变影响GABA 能神经元功能而导致癫痫[5-6]。关于SCN1A突变效应的研究显示除LOF致病外,功能获得性 (gain of function,GOF) 以及混合效应突变也可导致SCN1A谱系病[3]。SCN1A疾病谱系病包括严重的癫痫如Dravet 综合征、婴儿癫痫伴游走性局灶性发作、轻型自限性癫痫、药物反应性癫痫如全面性癫痫伴热性惊厥加症(GEFS+),还可伴多种类型的非痫性运动障碍[7-10]。本文患儿虽然为早发癫痫,但患儿无热敏感现象,癫痫发作对药物反应良好,分析可能的原因为该突变在通道的DI的S2区,为非电压感受区而导致较轻的癫痫表型,但该患儿伴有发育落后及难以控制的非痫性肌阵挛样发作,仍需继续延长随访时间以观察患儿病情进展。
对于常见的SCN1A突变导致癫痫一般不推荐使用Na+通道阻滞剂,因其可能加重癫痫发作。但研究发现基因突变效应即功能型(LOF、GOF及混合性突变)决定表型及对治疗反应,对于GOF患者,Na+通道治疗可能有效。目前已报道1 200多种SCN1A突变类型[11],在这些突变中观察到某些规律如截断性突变常产生LOF而导致Dravet综合征,但这些突变仍缺乏一对一基因型-表型关系,因为部分 LOF 也可导致良性倾向的癫痫如GEFS+[12]。而且,近年来发现即便SCN1A错义GOF突变也可导致比Dravet综合征更严重、早发的进行性癫痫性脑病,伴运动障碍包括舞蹈手足徐动、肌张力障碍及非癫痫性肌阵挛等,研究者认为符合上述表现者被命名为发育性癫痫-运动障碍性脑病[3,13]。本文患儿起病早,伴脑病表现,虽然癫痫控制较好,但运动障碍控制差,发育落后明显,符合发育性癫痫-运动障碍性脑病的表现。结合该患儿癫痫发作对奥卡西平治疗部分有效,提示该错义突变可能为GOF。既往关于SCN2A的研究提示,3个月内起病的患儿提示为GOF,对Na+通道阻滞剂有效,而起病年龄越晚则越可能是LOF[2,14]。目前关于SCN1A的研究还未发现类似规律,需在今后进一步加强对突变功能型的研究,以得出相应规律。
癫痫和阵发性运动障碍是由皮层、皮层-基底节连接或小脑功能短暂异常导致神经元网络障碍所致。这两种短暂性、发作性疾病通常由通道离子基因突变引起[1,15]。SCN1A除在皮层高表达外,在基底节及小脑也有较高表达,这种脑区表达特点进一步为SCN1A突变导致发育性癫痫-运动障碍性脑病提供了理论基础。除SCN1A外,目前还报道了多个基因突变可导致癫痫合并运动障碍,包括SCN8A、DEPDC5、SLC16A2、PNKD、KCNMA1、SLC2A1、SCN2A及ATP1A2等[1]。由于痫性发作与非痫性发作常同时在该类患者中发生,加之低龄儿童EEG特点,导致识图困难,进一步加剧了鉴别的难度。本文患儿早期的发作性症状为痫性发作,但却缺乏典型发作期图形,提醒临床医生对于低龄儿发作需结合患儿发作症状仔细反复读图,寻找发作前后轻微的EEG变化。
总之,SCN1A基因型-表型关系值得深入研究,发育性癫痫-运动障碍性脑病,一种更为严重的癫痫性脑病,可能是SCN1A突变的表型之一。对于早发非Dravet综合征型癫痫患者,可考虑尽早试用Na+通道阻滞剂治疗。
