SPAST基因新位点突变致遗传性痉挛性截瘫的家系报道
2022-09-20杨婷婷吴怀宽张冉冉方熙勤朱翠靳阳姜荆吴玉娇严翠华刘学伍
杨婷婷,吴怀宽,张冉冉,方熙勤,朱翠,靳阳,姜荆,吴玉娇,严翠华,刘学伍,3
遗传性痉挛性截瘫(hereditary spastic paraplegia,HSP)是一种临床和遗传异质性的神经系统退行性疾病,是以双下肢进行性痉挛性截瘫和肌无力为特征的综合征;HSP的遗传基础复杂,目前已发现80多个致病基因,涉及孟德尔遗传的所有模式[常染色体显性遗传(autosomal dominant ,AD)、常染色体隐性遗传(autosomal recessive, AR)、x连锁遗传(x-linked recessive,XR)及非孟德尔线粒体母系遗传(mitochondrial maternal inheritance)][1]。有研究表明,AD-HSP和AR-HSP的全球患病率大致相等[2]。也有研究表明不同地区、不同国家和不同种族之间发病率也有差异,在0.9/10万~10/10万之间,且男性多于女性[3]。SPAST基因突变导致的HSP 4型(SPG4)占所有HSP病例的25%以上和常染色体显性HSP患者的50%以上[4]。本文对1个SPAST基因位点首次突变的HSP家系的致病基因、临床特点及其家系情况进行分析及讨论,现将结果报道如下。
1 资料与方法
1.1 一般资料
先证者,女,46岁,因“双下肢无力、行走困难4年”就诊于山东大学齐鲁医院神经内科门诊,患者主要表现为行走缓慢,抬腿困难,剪刀步态,稍有跛行,偶有扑倒。查体:中年女性,颅神经未见异常,双上肢肌力及肌张力正常,肱二头肌反射、肱三头肌反射、桡骨膜反射亢进,双侧霍夫曼征阳性。双下肢近端、远端肌力正常,痉挛性肌张力增高,膝反射及踝反射亢进,双侧巴宾斯基征(+)、查多克征(+)、奥本海姆征(+)、戈登征(+),足下垂、弓形足畸形。余查体未见明显异常。辅助检查:头颅、全脊髓 MRI 未见异常,肌电图未见异常。现患者正在服用药物治疗:巴氯芬10 mg(3次/d)、申维30 mg(3次/d)及硫辛酸100 mg(3次/d)。先证者的母亲目前81岁,60多岁起病,临床表现主要为双下肢缓慢进展的痉挛、无力、步态异常,目前伴有尿失禁、双手动作笨拙、僵硬,已长期卧床、两腿僵直、弯曲费力、在扶持下都不能行走,目前生活已不能自理。先证者兄长与其存在相似的临床表现。先证者兄长的2个儿子与先证者的儿子临床症状较轻,主要表现为轻微剪刀步态,运动后感双下肢无力、运动耐受量下降、稍僵硬。该家系的临床症状与国内研究报道的SPG4家系的患者相似[3,5]。本研究患者及其家属已签署知情同意书。
1.2 方法
在取得患者及家属知情同意后,采集先证者外周静脉血2 mL,置于EDTA抗凝管中。 采用全血基因组DNA提取试剂盒(Beijing Precision Gene Technology Co., LTD)提取DNA,检测DNA浓度和纯度。使用神经系统捕获包检测+高通量测序的方法,检测患者基因层面的致病因素。将测序数据经美国国立生物技术信息中心数据库(NCBI)、千人基因组数据库(1000Genome)、基因组聚合数据库(gnomAD)及人类外显子组整合数据库(ExAC)过滤,结合 HGMD数据库筛选。
2 结果
先证者肱二头肌反射、肱三头肌反射、桡骨膜反射亢进,双侧霍夫曼征阳性。双下肢近端、远端肌力正常,痉挛性肌张力增高,膝反射及踝反射亢进,双侧巴宾斯基征(+)、查多克征(+)、奥本海姆征(+)、戈登征(+),足下垂、弓形足畸形(图1)。对该患者及其家属进行门诊问诊结果显示4代家系19名成员中,共有6名患者有临床症状(先证者、其母及其子已明确诊断,其余患者有临床症状,但未做基因检测),家系调查图谱详见图2。基因检测结果发现该患者在SPAST基因(NM_014946.3)第8外显子区域携带一处单杂合变异:c.1105A>C(腺嘌呤>胞嘧啶),导致氨基酸改变p.T369P(苏氨酸>脯氨酸)。后经sanger测序法验证,确定其为母源变异。根据美国大学医学遗传学(American College of Medical Genetics,ACMG)指南,该变异可评级为Likely pathogenic(疑似致病性变异),PM1+PM2+PP3+PP4。PM1:此变异位于关键功能域,上下游邻近位置均有致病变异报道;PM2:正常人群变异数据库未见报道;PP3:REVEL预测有害;PP4:表型与疾病符合。在基因检测结果提示先证者有基因突变(SPAST基因外显子测序质量报告见图3、基因读序比对见图4)后,经与先证者及其家属沟通决定对先证者的母亲及儿子进行基因检测。结果发现先证者的母亲及先证者儿子存在相同的杂合变异位点,且都有相应的临床症状,说明SPAST基因位点杂合突变可能为其家系发病原因。先证者、先证者母亲以及先证者儿子的基因检测结果见图5。

图1 先证者高弓足
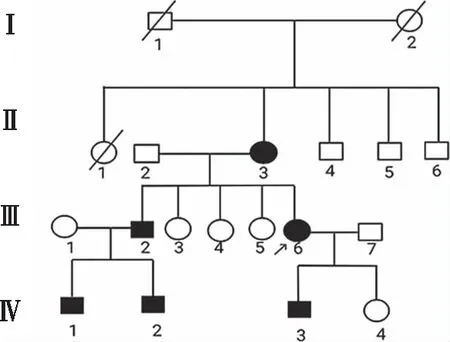
注:□为健康男性,○为健康女性,■为男患者,●为女患者,为先证者,/为死亡者。图2 HSP家系图

图3 先证者SPAST基因外显子测序质量报告图

图4 先证者SPAST基因读序比对图

注:DNA测序结果显示,先证者及母亲和先证者儿子具有相同的单杂合子变异,即SPAST c.1105A>Cchr2:32352023p.T369P。图5 先证者、先证者母亲及先证者儿子的基因检测结果
3 讨论
HSP可根据临床表型、遗传方式及病理生理分子机制进行分类,SPG4型由SPAST基因(2p22.3)杂合突变导致,该基因编码spastin蛋白,该蛋白由 616 个氨基酸组成,属于 ATP 酶(AAA)蛋白家族成员,SPAST基因突变可引起 spastin 蛋白功能异常,从而影响微管的切割及与微管相关的活动[6]。目前,SPAST基因的突变方式通常有错义突变、无义突变、剪切位点和缺失/插入的突变[7]。根据临床表型,可分为单纯型和复杂型;单纯型HSP主要以AD方式遗传,而复杂型的HSP主要以AR方式遗传[8]。HSP的单纯型临床特征为进行性步态异常、平衡障碍和下肢本体感觉减退等[9]。复杂型的HSP主要表现为神经症状和非神经症状,其中神经症状有小脑功能障碍、认知障碍及癫痫等[10];非神经症状主要包括眼科异常、脊柱侧弯、髋关节脱位及各种脚畸形等[11]。HSP的临床诊断取决于有无下肢进行性痉挛性偏瘫步态病史以及有无其他临床特征的家族史[6]。HSP分子诊断主要取决于区分导致该病的突变基因类型,患者的详细家族史和发展史在诊断中起关键作用。脑和脊髓的磁共振成像(MRI)可以帮助区分SPG类型[12]。由于HSP的高度临床异质性且各个亚型之间在临床表现上有很多的重叠,因此,基因检测对于HSP的诊断和分型很重要[13]。目前,对于HSP尚无特定的根治方法。但是一些对症治疗,如物理疗法、抗痉挛药(巴氯芬、替扎尼定、地西泮)、肉毒杆菌毒素及矫形器对于预防并发症如多发性挛缩、疼痛、骨折及提高患者的生活品质是很重要的[6]。在HSP-SPG4中,微管切断会导致部分基因功能丧失,因此可以通过基因治疗来纠正。另外,遗传检测和咨询可帮助患者和家属了解该疾病以及遗传给下一代的风险[14]。
本文先证者临床主要表现为行走缓慢,抬腿困难,剪刀步态,稍有跛行,偶有扑倒。查体:肱二头肌反射、肱三头肌反射、桡骨膜反射亢进,双侧霍夫曼征阳性。双下肢近端、远端肌力正常,痉挛性肌张力增高,膝反射及踝反射亢进,双侧巴宾斯基征(+)、查多克征征(+)、奥本海姆征(+)、戈登征(+),足下垂、弓形足畸形。HSP 4型常表现为单纯型。该患者虽有临床症状,但是头颅、全脊髓 MRI未见异常,考虑到患者发病时间较短,因此头颅、全脊髓MRI可正常,根据临床表现及基因检测结果,符合HSP 4型单纯型的诊断。基因检测结果提示先证者、其母及其子在SPAST基因(NM_014946.3)第8外显子区域携带一处单杂合变异:c.1105A>C(腺嘌呤>胞嘧啶),导致氨基酸改变p.T369P(苏氨酸>脯氨酸)。该突变位点于2014年由Ishiura等[15]报道,但在CNKI、万方数据知识服务平台、维普中文科技期刊数据库、PubMed查阅相关资料发现该突变位点在我国尚无报道。有研究表明,HSP的起病年龄跨度可从婴幼儿到老年 (70岁以上) ,主要取决于不同的基因型,即使在同一家系的同一突变中,不同的受累成员其发病年龄和疾病进展也不尽相同[16]。在本文先证者家系中,先证者的母亲在60多岁起病,先证者与其兄长都在40岁左右起病,先证者儿子及先证者兄长的2个儿子在20岁左右起病,故该家系起病情况符合HSP临床表现的异质性。本研究的局限性在于未能对有临床症状的另外3位患者进行基因检测以进一步确诊,但是在该家系中已有3位患者行基因检测并且有相应的临床症状。故SPAST基因(NM_014946.3)第8外显子区域携带一处单杂合变异:c.1105A>C(腺嘌呤>胞嘧啶),导致氨基酸改变p.T369P(苏氨酸>脯氨酸)可能为该家系的致病基因。
HSP突变的基因位点较多且临床症状多样,诊断需结合临床表现及辅助检查,其中基因检测尤为重要。通过基因检测明确诊断后,可给予相应的对症治疗,减轻症状并延缓疾病进展。另外,可通过医学辅助生殖技术筛选优质基因,帮助患者孕育健康下一代,以减少或终止HSP的遗传。
