Gd对Mg-xGd-1Er-1Zn-0.6Zr合金显微组织和腐蚀行为的影响
2022-09-19刘雄飞杜文博付军健王云峰李淑波朱训明王朝辉
刘雄飞,杜文博*,付军健,王云峰,李淑波,朱训明,王朝辉
(1 北京工业大学 材料与制造学部,北京 100124;2 威海万丰镁业科技发展有限公司,山东 威海 264209)
镁及其合金具有密度低、比强度与比刚度高、易切削加工以及可回收等优点,被誉为“21世纪绿色工程材料”,在汽车行业、3C电子产品和航空航天等领域有着广泛的应用前景[1]。但纯镁的强度较低,其抗拉强度仅为40 MPa,无法作为结构材料获得应用[2]。此外,由于镁标准电极电位较低(-2.37 V),极易发生腐蚀,例如,Zhao等[3]发现纯镁在3.0%(质量分数,下同)NaCl溶液中的腐蚀速率达到2.7 mm/a。为进一步提高纯镁的力学性能和耐腐蚀性能,合金化是一条有效途径,通过合金元素的固溶或时效析出第二相,不仅可以有效提高镁合金的力学性能,同时也能改善镁合金的耐腐蚀性能。例如,AZ91合金[4-5],因其沿晶界连续均匀分布的β-Mg17Al12相,使得其力学性能和耐腐蚀性能均获得较大提升,铸态抗拉强度达到159 MPa,在3.5%NaCl溶液中的腐蚀速率为1.1 mm/a。除Al,Zn等合金化元素外,添加RE元素也可有效提升镁合金的力学性能和耐腐蚀性能。Li等[6]研究发现Mg-10Gd-4Y-0.3Zr合金峰时效后的抗拉强度达到325 MPa。Liang等[7]研究表明Mg-7Gd-3Y-0.4Zr合金经固溶处理后,在5.0%NaCl溶液中的腐蚀速率仅为0.5 mm/a。
近年来,含有长周期堆垛有序(long period stacking ordered,LPSO)相的稀土镁合金因其优异的力学性能受到人们广泛关注。Wang等[8]研究发现Mg-4Y-2Er-2Zn-0.6Zr合金固溶处理后,层片状的14H-LPSO相转变为块状的18R-LPSO相,其抗拉强度达到215 MPa,伸长率为21%。Ramezani等[9]研究表明Mg-8Gd-4Y-2Zn-0.6Zr合金经多向锻造后,获得超细晶组织及高体积分数的LPSO相,其抗拉强度达到581 MPa,伸长率为16%。虽然含LPSO相的稀土镁合金力学性能因LPSO相得到显著改善,但LPSO相对稀土镁合金耐腐蚀性能的影响一直存在争议。Zhang等[10]认为铸态Mg-7Y-2Zn合金晶界处分布的连续块状LPSO相起到了腐蚀屏障的作用,合金在3.5%NaCl溶液中的腐蚀速率为2.3 mm/a。Bao等[11]认为铸态Mg-9Y-3Zn-0.5Zr合金中致密的块状LPSO相能阻止腐蚀向基体扩张,使合金在3.5%NaCl溶液中的腐蚀速率控制在6.8 mm/a。然而,Zhang等[12]研究了铸态Mg-11Gd-3Y-5Zn合金,发现晶界处不连续分布的LPSO相因与镁基体电位差较大,充当强烈的阴极相加速腐蚀,在3.0%NaCl溶液中腐蚀速率达到756.8 mm/a。Bi等[13]研究发现Mg-12Dy-1Zn合金经过固溶处理后,晶粒内部析出大量层片状的14H-LPSO相,在3.5%NaCl溶液中腐蚀速率达到267.7 mm/a。
为进一步分析LPSO相对稀土镁合金的腐蚀行为的影响机制,本工作针对前期开发的Mg-Gd-Er-Zn-Zr高强镁合金[14-15],通过改变Gd的含量,调控第二相数量和分布,研究第二相对合金腐蚀行为的影响规律,为阐明含LPSO相的Mg-RE合金的腐蚀机理提供理论依据。
1 实验材料与方法
采用纯Mg(99.99%),纯Zn(99.99%),Mg-30%Gd,Mg-20%Er和Mg-30%Zr中间合金作为原材料。合金在井式电阻炉中进行熔炼,采用N2和SF6的混合气体(体积比为20∶1)进行保护。熔体温度为720 ℃时进行浇铸,获得铸态合金。合金成分采用XRF-1800型X射线荧光光谱仪进行测试,结果如表1所示,三种合金中的实际Gd含量分别为6.66%,8.80%和10.76%,分别标记为合金A、合金B和合金C。采用D/MAX-3C型X射线衍射仪检测合金和腐蚀产物的物相;采用Axio imager A2m金相显微镜(OM)和Quanta650型扫描电镜(SEM)观察试样的显微组织与表面形貌,并利用SEM设备上带有的能谱仪(EDS)对试样表面进行元素分析;采用Image J软件分析试样的显微组织图片,统计第二相体积分数和晶粒尺寸。
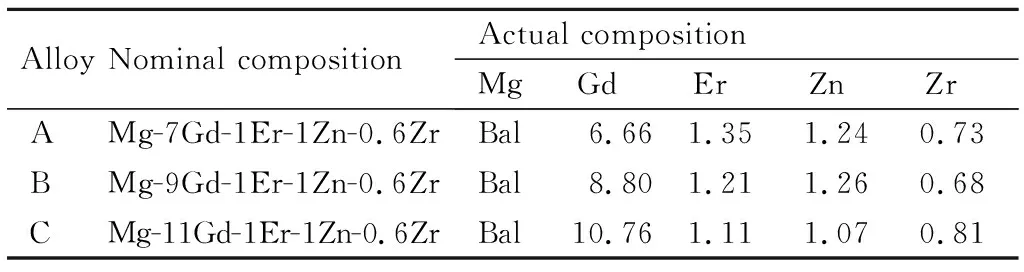
表1 三种合金的名义成分与实际成分(质量分数/%)Table 1 Nominal and actual compositions of three alloys (mass fraction/%)
在合金中间取样,切成10 mm×10 mm×8 mm的试样。将试样悬挂浸泡在3.5%NaCl溶液中,溶液体积与试样表面积比例为20∶1,浸泡不同时间后用铬酸溶液清除腐蚀产物,计算试样浸泡后的失重腐蚀速率。浸泡过程始终使用水浴保持温度为25 ℃。
采用Autolab电化学工作站测试试样的电化学性能,其中待测样品为工作电极,铂片为对电极(CE),与合金形成回路,饱和甘汞电极(SCE)为参比电极。在频率为0.01 Hz~100 kHz范围内进行电化学阻抗(EIS)测试,使用振幅为10 mV的正弦电压,采用ZSimDemo3.30d软件对电化学阻抗结果进行电路拟合。动电位极化测试的电压范围相对于开路电位±300 mV,测试是由负电位到正电位逐步扫描,扫描速度为0.5 mV/s。
2 实验结果和分析
2.1 显微组织
图1为铸态Mg-xGd-1Er-1Zn-0.6Zr合金的XRD图谱,结果显示这三种合金都是由α-Mg,Mg3Gd相和LPSO相组成。图2为铸态Mg-xGd-1Er-1Zn-0.6Zr合金的OM图,由图2可见,树枝状的黑色共晶相、灰色块状相和细小层状相沿晶界分布,且随Gd含量从7%增至11%,共晶相由不连续分布转变为半连续分布,层状相数量增多且贯穿晶粒内部,块状相逐渐消失。随着Gd含量从7%增至11%,合金晶粒尺寸减小,从46.1 μm降至39.0 μm,但第二相的体积分数增加,共晶相的体积分数从1.9%增至5.2%,层状相的体积分数从11.7%增至26.7%。
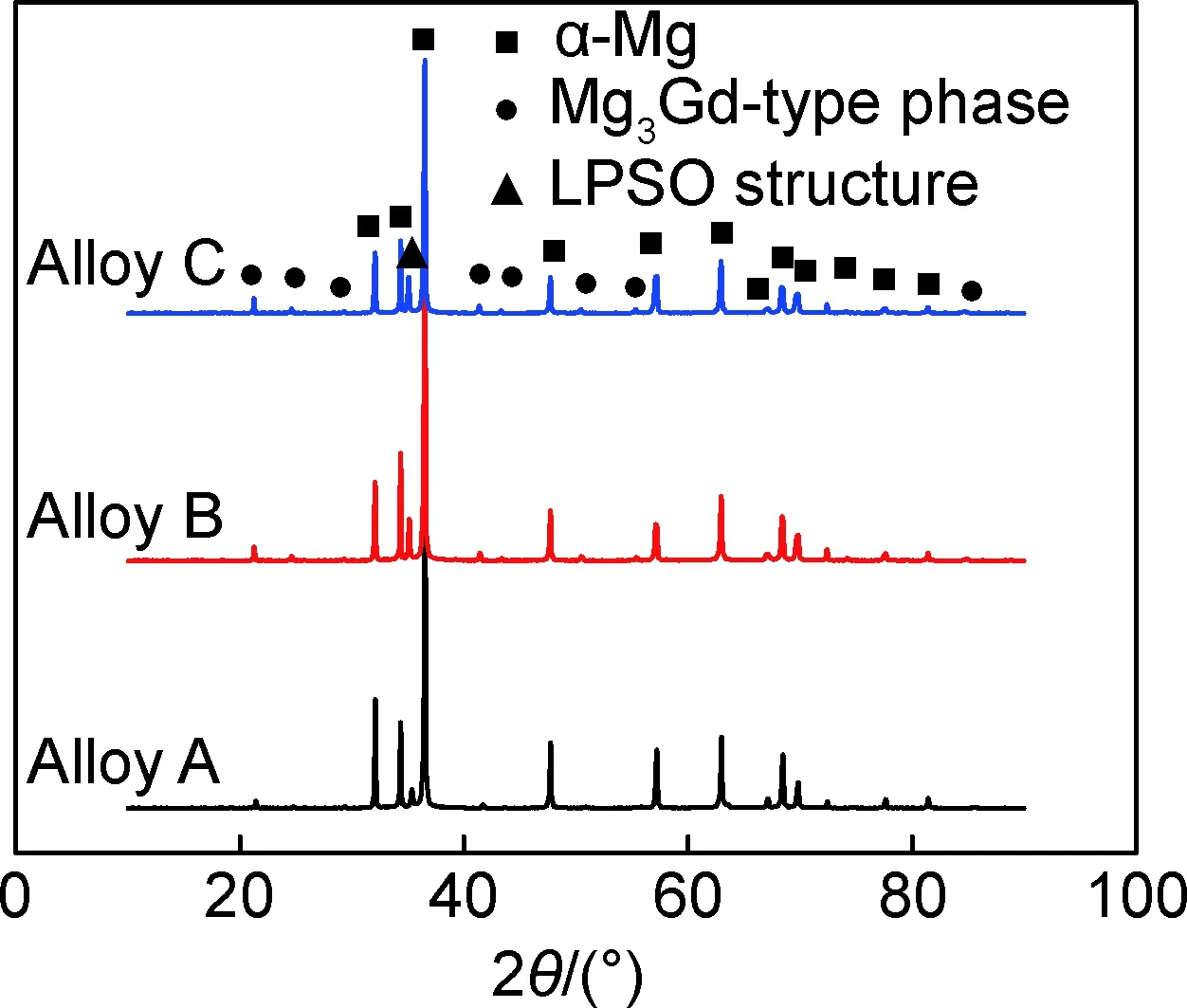
图1 铸态Mg-xGd-1Er-1Zn-0.6Zr合金的X射线衍射图谱Fig.1 XRD patterns of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys

图2 铸态Mg-xGd-1Er-1Zn-0.6Zr合金光学显微组织(a)合金A;(b)合金B;(c)合金CFig.2 Optical microstructures of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys(a)alloy A;(b)alloy B;(c)alloy C
图3为铸态Mg-xGd-1Er-1Zn-0.6Zr合金的SEM图,图中各点对应的EDS分析结果如表2所示。随着Gd含量从7%增至11%,α-Mg内固溶的Gd元素含量从0.20%增至0.60%,Zn元素含量从0.02%增至0.12%,溶质原子溶入基体中能改善基体的表面电位且在基体表面形成致密的氧化膜[16]。合金内共晶相元素含量相同,(Mg,Zn)与(Gd,Er)元素的原子比均接近于3,结合XRD图谱,可以确定共晶相为(Mg,Zn)3(Gd,Er)相。块状相中的元素成分接近Mg97Zn1(Gd,Er)2,与文献报道[17]的LPSO结构成分范围相近,结合XRD图谱可以确定其为LPSO结构。层状相主要含有Zn和RE(Gd,Er)元素,结合Jia等[14]的研究结果,确定其为几个纳米宽的LPSO结构。

图3 铸态Mg-xGd-1Er-1Zn-0.6Zr合金的SEM图(a)合金A;(b)合金B;(c)合金CFig.3 SEM images of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys(a)alloy A;(b)alloy B;(c)alloy C
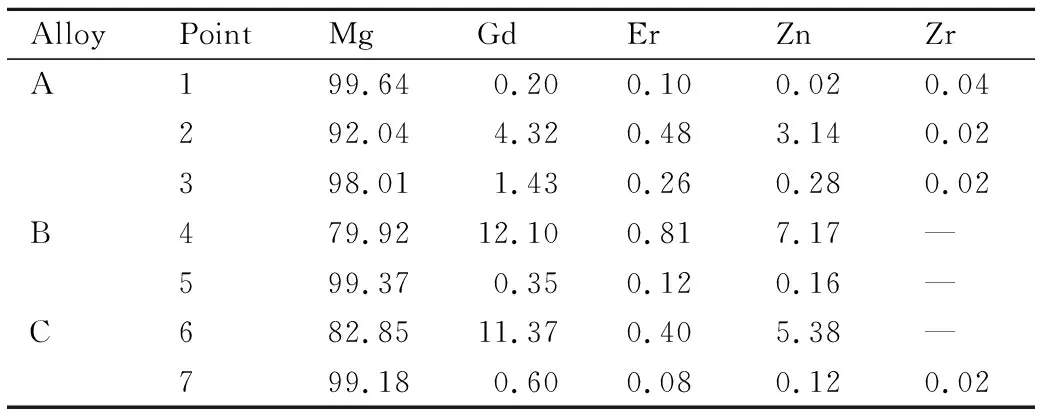
表2 图3各点对应的EDS结果(原子分数/%)Table 2 EDS results of the various points shown in fig.3 (atom fraction/%)
2.2 电化学实验
图4为铸态Mg-xGd-1Er-1Zn-0.6Zr合金在3.5%NaCl溶液中开路电位(OCP)的变化曲线。三种合金开路电位的变化都可分为3个阶段,A阶段表示开路电位迅速上升,B阶段表示开路电位缓慢上升,C阶段表示开路电位趋于稳定。开路电位的增加表明合金表面逐渐形成氧化膜,开路电位的降低表示合金表面的保护膜遭到破坏,开路电位保持相对稳定则代表腐蚀的进行与腐蚀产物的沉积达到了相对稳定的状态[18]。如图4所示,在A阶段和B阶段,三种合金的开路电位都随着浸泡时间的延长逐渐上升,这是由于表面形成了保护氧化膜,随着时间的延长,到达了C阶段,腐蚀进行与腐蚀产物积累达到了动态平衡,这时开路电位保持相对稳定。开路电位图中出现峰值的时间越早,代表局部腐蚀的潜伏期越短,表明局部腐蚀与腐蚀产物沉积之间的平衡越快[19],由图4可见,合金A出现峰值的时间最长,为1609 s,合金C出现峰值的时间最短,为851 s。这表明合金C局部腐蚀的活化期较短,较容易被腐蚀,这可能是由于其存在较多的析出相。

图4 铸态Mg-xGd-1Er-1Zn-0.6Zr合金在3.5%NaCl溶液中的开路电位变化Fig.4 Variation of the open circuit potentials of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys during immersion in 3.5%NaCl solution
图5为铸态Mg-xGd-1Er-1Zn-0.6Zr合金在3.5%NaCl溶液中开路电位稳定后所测的电化学阻抗(EIS)曲线。根据开路电位测试,每个单独的EIS测量都大约需要1800 s,而且整个测试过程都要处于开路电位稳定状态。如图5(a)所示,合金A的Nyquist图由一个高频容抗弧和一个低频容抗弧组成,合金B和合金C的Nyquist图都由一个高频容抗弧和一个低频感抗弧组成。高频容抗弧反映材料基体的电荷转移反应,低频容抗弧反映电荷或物质通过表面氧化层的传输过程,低频感抗弧的存在可归因于局部腐蚀起始阶段的腐蚀形核,可能是由于腐蚀介质中Cl-的存在使得表面膜更加活跃,低频感抗弧的存在表明表面膜的保护能力不足,基体已经开始腐蚀[20]。高频容抗弧越大,电荷穿过合金腐蚀产物膜与溶液之间的双电层越困难,高频容抗弧的直径就等于工作电极的电荷转移电阻,因而高频容抗弧的大小可以反映耐蚀性的好坏[21],由Nyquist图可见,合金A的高频容抗弧最大,由此可判断合金A耐蚀性最好。由Bode图(图5(b))可见,三种合金从高频到低频区域,阻抗值都一直上升,低频区的模数值体现了合金的耐蚀性,结果显示合金A在低频区的模数值最大,表明其腐蚀倾向性最小。且由Bode图中的相位变化可知,三种合金都在高频区存在一个波峰,而在Bode图中最大相位角越大,对应的频率越低,代表合金的耐蚀性越好,结果显示合金A的峰值最大,表明其耐腐蚀性能最好。采用电阻、电容和电感等电学元件构建等效电路对EIS图谱进行进一步分析。图6为铸态Mg-xGd-1Er-1Zn-0.6Zr合金EIS阻抗谱的等效电路,图6(a)用于拟合合金A的阻抗响应,图6(b)用于拟合合金B和合金C的阻抗响应。图中Rs代表溶液电阻;Rct代表电荷转移电阻;CPEdl是恒相位元件,与电解质溶液和镁基体界面的双电层有关,用来代替理想的电容器来补偿系统中的非均匀性[22];Rf表示薄膜电阻;Cf表示薄膜电容;L表示电感;RL表示电阻。Rct和CPEdl的并联回路用来描述高频电容弧,较高的Rct值代表镁基体的溶解速率较低。Rf和Cf的并联回路用来描述低频电容弧,低频电容弧的存在可归因于合金表面氧化膜的存在[23]。RL和L的串联回路用来描述低频电感弧,低频电感弧的出现是由开始发生局部腐蚀所引发,合金B和合金C存在低频电感回路表示其耐蚀性较低。表3为利用ZsimDemo3.30d软件进行拟合后得到的数据,其拟合误差均在10-3数量级,表3显示,三种合金的电荷转移电阻分别是588.50,322.70,31.91 Ω·cm2,其数值越小,代表镁基体溶解速率越大,合金C数值最小,代表其耐腐蚀性最差。
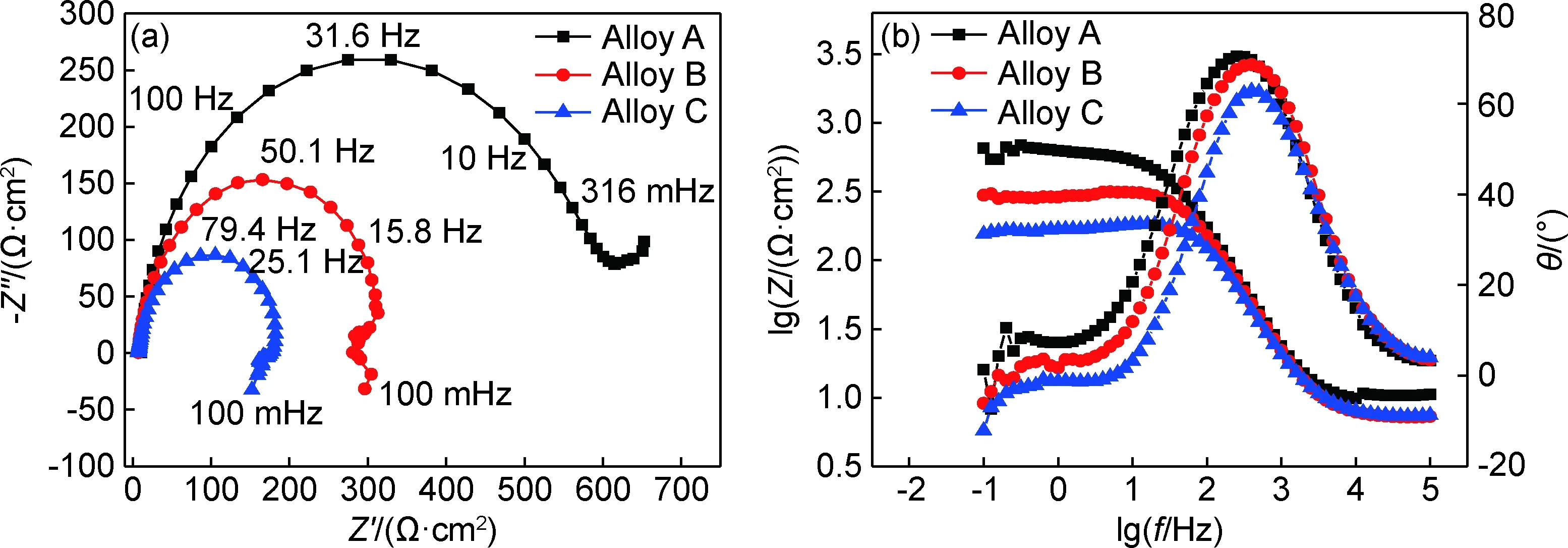
图5 铸态Mg-xGd-1Er-1Zn-0.6Zr合金在3.5%NaCl溶液中的电化学阻抗谱(a)Nyquist图;(b)Bode图Fig.5 Electrochemical impedance spectra of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys in 3.5%NaCl solution(a)Nyquist diagram;(b)Bode diagram

图6 铸态Mg-xGd-1Er-1Zn-0.6Zr合金电化学阻抗谱的等效电路(a)合金A;(b)合金B和合金CFig.6 Equivalent circuit models used for fitting the impedance spectra of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys(a)alloy A;(b)alloy B and alloy C
图7为铸态Mg-xGd-1Er-1Zn-0.6Zr合金的动态电位极化曲线,其中阳极分支(anodic branches)代表金属阳极的溶解,阴极分支(cathodic branches)代表阴极析氢反应。由阳极分支可知,合金A更加陡峭,表示极化程度较大,耐蚀性较好[24]。由阴极分支可知,在电位比临界点蚀电位更负时表现出线性Tafel特性,即存在Tafel线性区,且相比阳极分支更加陡峭,这表示阴极反应控制了腐蚀过程,且合金A的氢过电位最大,代表其耐蚀性最好。表4为Tafel外推法得到的铸态Mg-xGd-1Er-1Zn-0.6Zr合金的电化学参数,由表4可见,三种合金的腐蚀电流密度分别为2.21×10-5,3.76×10-5,3.97 ×10-5A/cm2,合金A的腐蚀电流密度最小,且其极化电阻最大,约为2016.30 Ω,代表合金A的耐腐蚀性能最好。

表3 铸态Mg-xGd-1Er-1Zn-0.6Zr合金电化学阻抗谱拟合结果Table 3 Fitting results of the EIS for as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys
2.3 腐蚀形貌
图8为合金A在3.5%NaCl溶液中浸泡后带有腐蚀产物的表面形貌图。由图8可见,浸泡1 h后,合金表面开始出现黄色膜层和黑色丝状腐蚀产物,随着浸泡时间的延长,表面的黑色丝状腐蚀产物逐渐扩展,浸泡14 h后,合金表面已失去金属光泽,表面覆盖着灰白色的腐蚀产物层。
图9为合金A在3.5%NaCl溶液中浸泡后带有腐蚀产物的SEM图,图中各点对应的EDS分析结果如表5所示。浸泡1 h时,合金表面生成了针叶状和花瓣状的腐蚀产物,主要含有Mg,O和少量的稀土元素Gd和Er,且花瓣状腐蚀产物中O元素含量较多,结合图8可知,合金表面生成的黄色膜层是由针叶状的腐蚀产物组成,黑色丝状腐蚀产物是由花瓣状产物连接形成。浸泡4 h后,针叶状的腐蚀产物尺寸增加,花瓣状的腐蚀产物逐渐堆积在合金表面,形成团絮状,其中O和Mg元素的原子比接近于2,结合XRD图谱,可确定其为Mg(OH)2。
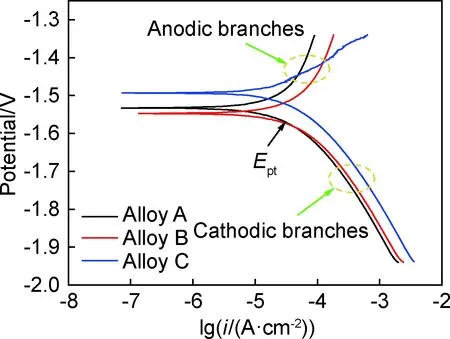
图7 铸态Mg-xGd-1Er-1Zn-0.6Zr合金的动态电位极化曲线Fig.7 Potentiodynamic polarization curves of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys in 3.5%NaCl solution

表4 铸态Mg-xGd-1Er-1Zn-0.6Zr合金的电化学参数Table 4 Electrochemical parameters of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys
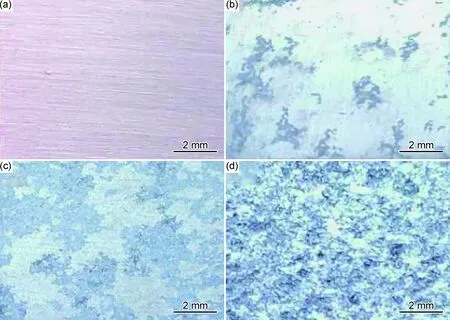
图8 合金A在3.5%NaCl溶液中浸泡不同时间后带有腐蚀产物的表面形貌(a)0 h;(b)1 h;(c)4 h;(d)14 hFig.8 Surface morphologies of alloy A after immersion in 3.5%NaCl solution for different time with corrosion products(a)0 h;(b)1 h;(c)4 h;(d)14 h
图10为铸态Mg-xGd-1Er-1Zn-0.6Zr合金在3.5%NaCl溶液中浸泡8 h后的腐蚀产物XRD图谱。结果表明,除了镁基体以外,合金的腐蚀产物主要是由Mg(OH)2组成。从XRD图谱中并没有发现Zn,RE元素的氧化物以及β-(Mg,Zn)3(Gd,Er)和LPSO析出相,这说明合金在3.5%NaCl溶液中浸泡后,第二相自身没有被腐蚀,但因其周围的镁基体被腐蚀后而发生脱落,部分进入溶液中,而残留的第二相过少,未能被检出[25-26]。且随着Gd含量从7%增至11%,镁基体的衍射峰强度降低,Mg(OH)2的衍射峰强度升高,说明合金C表面的腐蚀产物较多。

图9 合金A在3.5%NaCl溶液中浸泡不同时间后带有腐蚀产物的SEM图(a)1 h;(b)4 hFig.9 SEM images of alloy A surfaces after immersion in 3.5%NaCl solution for different time with corrosion products(a)1 h;(b)4 h
图11为铸态Mg-xGd-1Er-1Zn-0.6Zr合金浸泡在3.5%NaCl溶液中去掉腐蚀产物后的SEM图。由图11可知,浸泡1 h后,晶界处最先开始腐蚀且出现宽度为5~27 μm的丝状腐蚀坑,这是由于分布在晶界处的析出相因其电位较高充当阴极相,加速周围镁基体的腐蚀,因而晶界处的镁基体最先开始腐蚀,当析出相周围的镁基体被腐蚀后,析出相发生脱落[27]。随着浸泡时间的延长,腐蚀从晶界处向晶粒内部扩展,浸泡14 h后,合金A晶粒内部产生直径约4 μm的点蚀坑,合金B表面出现直径43~90 μm的巨大坑洞,合金C表面则出现数量较多的点蚀坑,直径从2~15 μm不等。合金B表面的坑洞可能是由晶粒内部的点蚀坑扩展连接而形成。浸泡24 h后,腐蚀纵向扩展,留下较深的孔洞,合金A表面点蚀坑直径增至15 μm,合金B表面出现大量纤维状区域,合金C表面出现凹坑以及裂纹,纤维状区域是由于析氢反应所产生的气孔连接而形成[28],表明合金B析氢反应剧烈,腐蚀严重。
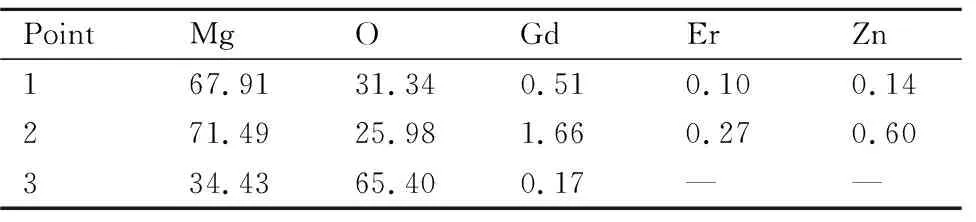
表5 图9各点对应的EDS结果(原子分数/%)Table 5 EDS results of the various points shown in fig.9 (atom fraction/%)
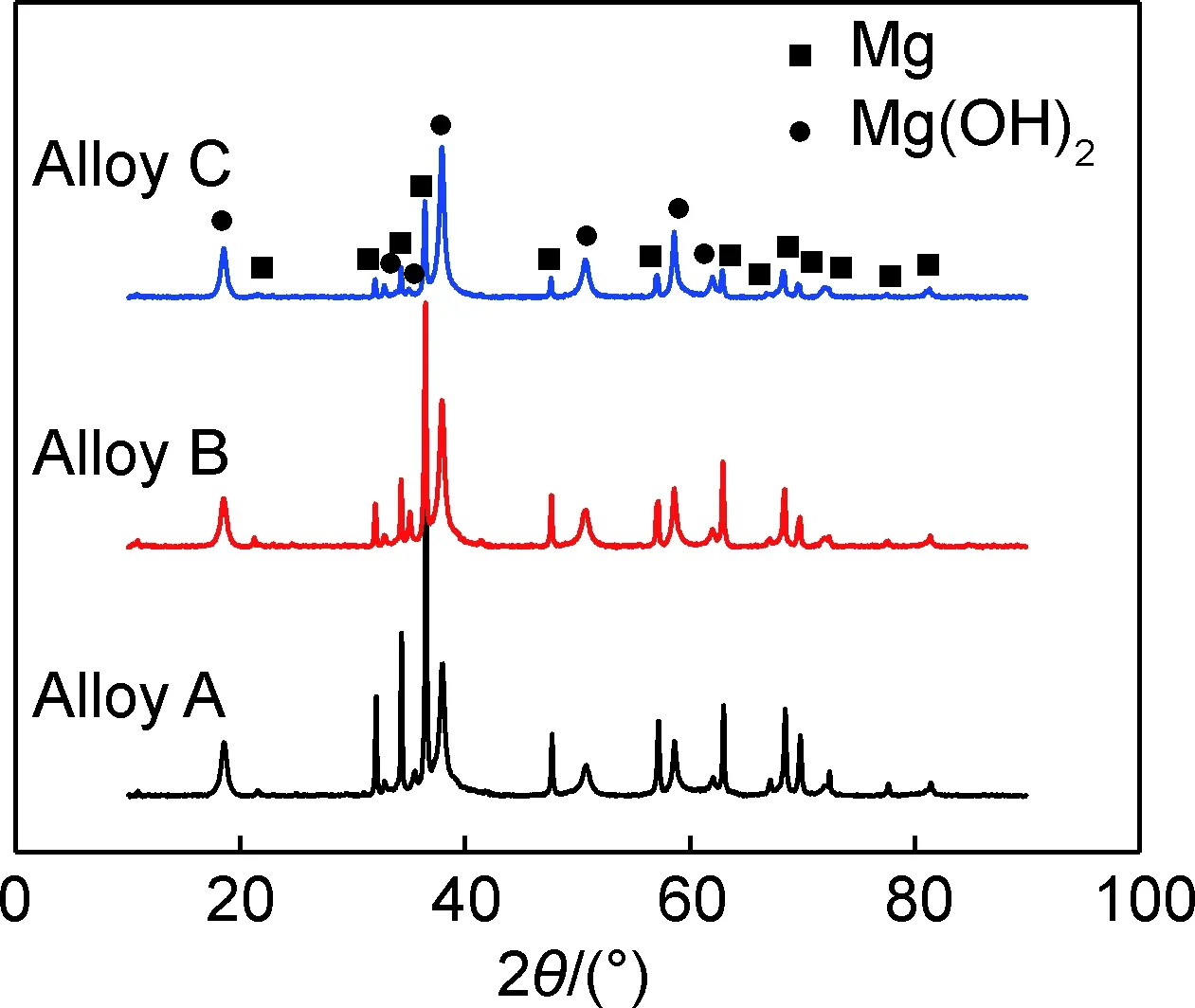
图10 铸态Mg-xGd-1Er-1Zn-0.6Zr合金在3.5%NaCl溶液中浸泡8 h后腐蚀产物的XRD谱图Fig.10 XRD patterns of corrosion products of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys after immersion in 3.5%NaCl solution for 8 h
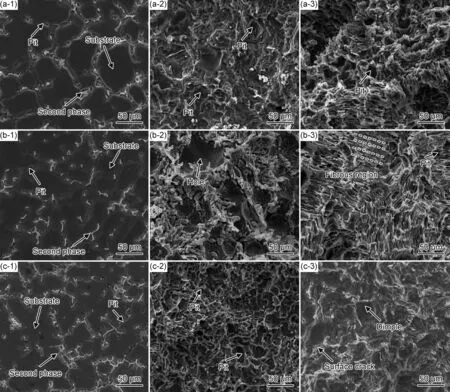
图11 铸态Mg-xGd-1Er-1Zn-0.6Zr合金在3.5%NaCl溶液中浸泡不同时间后去掉腐蚀产物的腐蚀形貌(a)合金A;(b)合金B;(c)合金C;(1)1 h;(2)14 h;(3)24 hFig.11 Morphologies of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys after immersion in 3.5%NaCl solution for different time without corrosion products (a)alloy A;(b)alloy B;(c)alloy C;(1)1 h;(2)14 h;(3)24 h
2.4 腐蚀速率
图12为采用失重法测得的铸态Mg-xGd-1Er-1Zn-0.6Zr合金的腐蚀速率。由图12可知,合金的腐蚀速率都随浸泡时间的延长而呈现增加的趋势,经14 h浸泡后合金B的失重腐蚀速率更高。在浸泡初期,合金A的失重腐蚀速率更低,三种合金的腐蚀速率变化较缓慢,这是由于浸泡前期产生的腐蚀产物膜对合金有一定的保护作用。随着浸泡时间的延长,Cl-不断渗入腐蚀产物膜中,合金的失重腐蚀速率逐渐上升。浸泡14 h后,合金C的腐蚀速率增长减缓,低于合金B的腐蚀速率,这可能是因为合金C晶粒内部分布着的细小层状LPSO相在一定程度上具有腐蚀屏障的作用,阻止腐蚀扩展,使得腐蚀速率增长减缓。在3.5%NaCl溶液浸泡实验表明,铸态合金的腐蚀速率顺序如下:合金C>合金B>合金A。

图12 铸态Mg-xGd-1Er-1Zn-0.6Zr合金失重腐蚀速率Fig.12 Corrosion rate obtained by mass loss of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys
图13为铸态Mg-xGd-1Er-1Zn-0.6Zr合金的腐蚀机理示意图。在腐蚀初期,晶界处分布着β-(Mg,Zn)3(Gd,Er)相、块状LPSO相和细小层片状LPSO相,因其相对于镁基体电位较高,充当阴极加速周围镁基体腐蚀,腐蚀最先开始于晶界附近,呈丝状腐蚀的特征,部分第二相因周围镁基体腐蚀而脱落并溶入3.5%NaCl溶液中,残留析出相继续加速周围镁基体腐蚀。随着浸泡时间的延长,合金表面开始形成氧化膜,根据图10的XRD结果可知,氧化膜的主要成分是Mg(OH)2,而Mg(OH)2疏松多孔,无法对基体起到良好的保护作用。因而,Cl-不断渗入氧化膜,加速腐蚀扩展,腐蚀从晶界处开始向晶粒内部蔓延,出现如图11(c-2)所示大量的点蚀坑,点蚀坑逐渐扩展连接,形成了较大的坑洞。
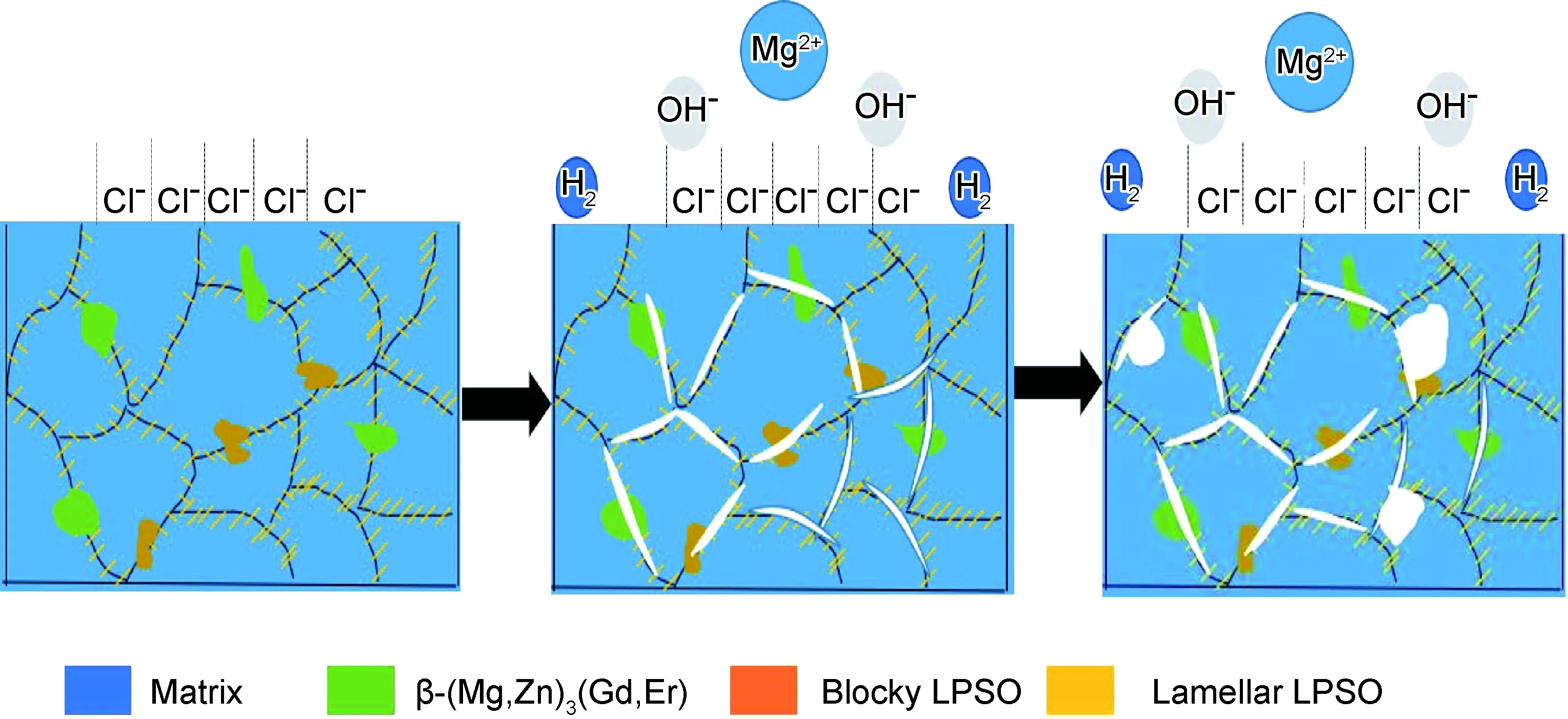
图13 铸态Mg-xGd-1Er-1Zn-0.6Zr合金随浸泡时间延长的腐蚀机理示意图Fig.13 Schematic diagrams of the corrosion mechanism with immersion time of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys
合金A中第二相含量较少,微电偶腐蚀效应较弱,因而其腐蚀速率较低,合金B中第二相含量增加,微电偶腐蚀效应加强,导致腐蚀速率增大,且析氢反应剧烈,产生大量气孔,形成纤维状区域。合金C中β-(Mg,Zn)3(Gd,Er)相体积分数增至5.2%,并从沿晶界不连续分布转变为半连续分布,层片状LPSO相体积分数增至26.7%,并沿着晶界贯穿晶粒内部,β-(Mg,Zn)3(Gd,Er)相和层片状LPSO相体积分数的增加,导致合金耐腐蚀性能下降,但大量细小层状LPSO相能阻止腐蚀扩展,使得合金C在8~24 h内腐蚀速率增长减缓。
3 结论
(1)铸态Mg-xGd-1Er-1Zn-0.6Zr合金中的析出相都沿着晶界分布,但当Gd含量从7%增至11%,合金析出相数量和分布都发生变化,β-(Mg,Zn)3(Gd,Er)相体积分数由1.9%增至5.2%,由不连续分布转变为半连续分布。块状LPSO相逐渐消失,层片状LPSO相体积分数由11.7%增至26.7%,且沿着晶界贯穿晶粒内部。
(2)开路电位测试表明,Gd含量较低的合金A到达峰值的时间最长,代表其局部腐蚀潜伏期较长,合金耐蚀性较好。电化学阻抗测试表明,Gd含量较高的合金C的高频电容回路弧度最小,代表其耐蚀性较差。
(3)在3.5%NaCl溶液浸泡实验表明,铸态Mg-xGd-1Er-1Zn-0.6Zr合金的腐蚀速率顺序如下:合金C>合金B>合金A。即随着Gd含量的增加,合金耐蚀性下降,这主要归因于第二相微电偶腐蚀效应和腐蚀屏障效应的共同作用。但大量细小的层状LPSO相也能阻止腐蚀扩展,使得合金C在8~24 h腐蚀速率增长减缓。
