荧光1,2,4-噻二唑-3,5-二酮衍生物的合成及其对精子的标记作用
2022-09-03姜兵兵王威威杨依婷李玉华邵志宇
姜兵兵, 王威威, 杨依婷, 李玉华, 刁 华*, 邵志宇*
(1. 东华大学 化学化工与生物工程学院,上海 201620; 2. 复旦大学 基础医学院 生殖与发育研究院,上海 200032; 3. 上海市生物医药技术研究院 国家卫生健康委员会计划生育药具重点实验室,上海 200032)
1,2,4-噻二唑-3,5-二酮类化合物是以噻二唑烷酮(TDZD)为核心骨架,改变2,4-位氮上连接的取代基而衍生出的一类氮硫杂环化合物。噻二唑烷酮类化合物可以作为药物分子设计中的先导化合物[1]。目前,市售药物分子中大多含有噻二唑烷酮结构。含有噻二唑烷酮骨架的化合物具有许多生物和药理活性,可作为糖原合酶激酶GSK-3β[2-5]、金黄色葡萄球菌(SrtA)[6]、抑制结肠炎[7]、抗菌活性[8]、抑制丙氨酸消旋酶[9]、抑制HL60细胞增殖[10]的抑制剂。其中,GSK-3β是一种普遍存在的丝氨酸/苏氨酸激酶,与多种重要的细胞功能有关,而该酶的抑制剂可能具有抗白血病、治疗Ⅱ型糖尿病和躁狂抑郁症等疾病的活性[2-5]。
通过高通量筛选,研究人员发现含1,2,4-噻二唑-3,5-二酮骨架的化合物对精子的活性具有抑制作用[11]。部分具有噻二唑烷酮骨架的化合物比传统的杀精剂壬苯醇醚-9效率更高,体细胞毒性更低。例如Tideglusib具有开发为新一代安全杀精避孕药物的潜力[11],但其具体作用靶点和分子作用机制还未知。本课题组前期设计并合成出了一系列含有噻二唑烷酮骨架的化合物,研究了其对精子活性的抑制作用,结果发现,含有噻二唑二酮五元环骨架的化合物对精子运动活性有一定的抑制作用,但还未能明确该类化合物与精子作用的具体位点。基于课题组前期工作[12],为了探究噻二唑二酮类化合物抑制精子活性的具体生物靶点,并揭示其抑制精子活性的机理,本文设计了一类带有荧光标记的活性噻二唑二酮化合物,并研究了其与精子的相互作用模式。含荧光基团的1,2,4-噻二唑-3,5-二酮衍生物的合成方法如下:(1)噻二唑二酮五元环骨架的构建(Scheme 1)[13]; (2)带有荧光基团的化合物的合成(Scheme 2)[14-15]; (3)将荧光化合物连接到噻二唑二酮骨架上,合成目标化合物S1~S3(Scheme 3)。
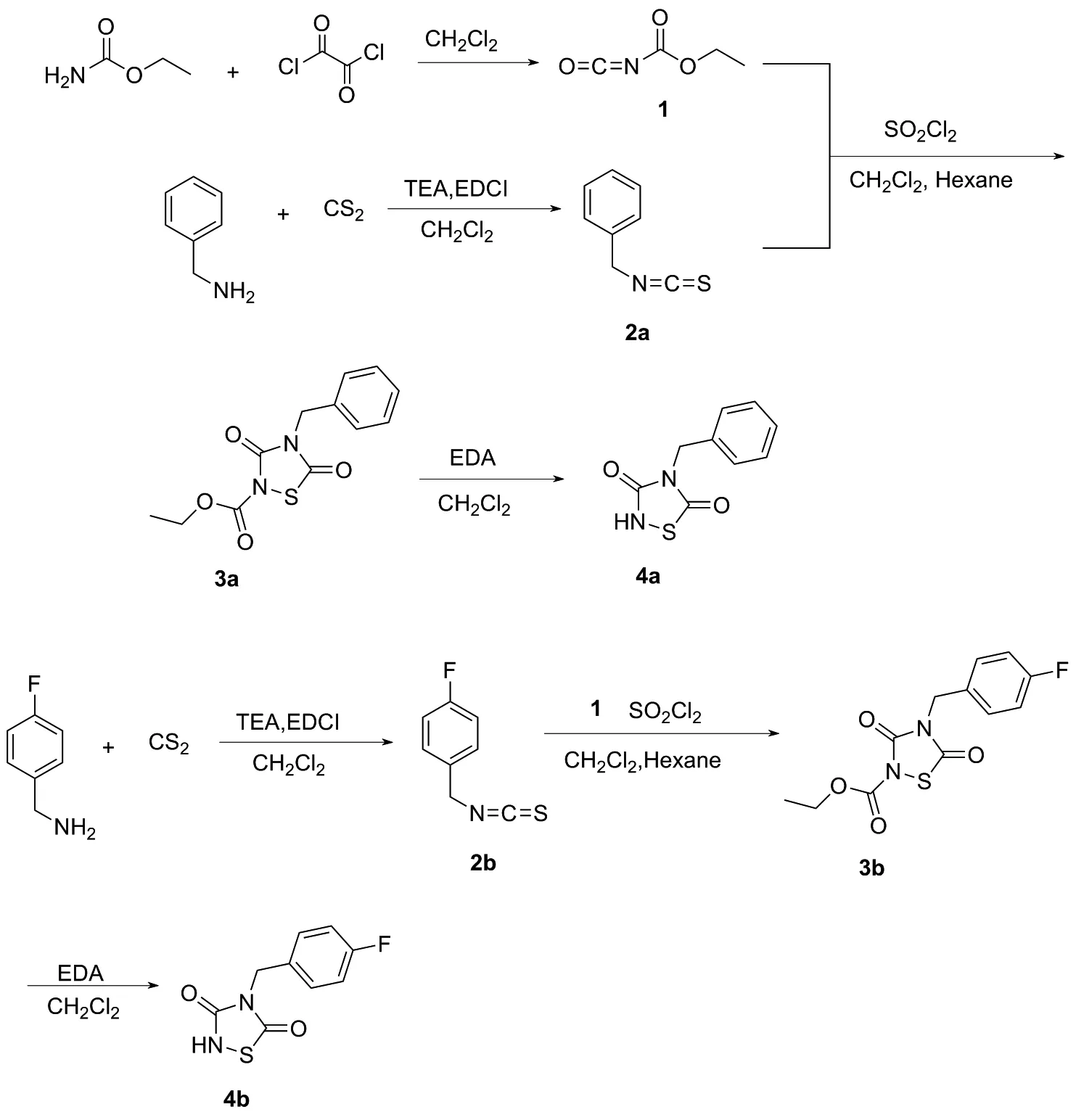
Scheme 1
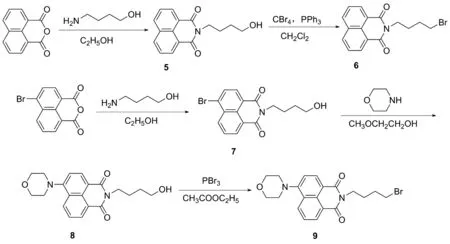
Scheme 2

Scheme 3
1 实验部分
1.1 仪器与试剂
Bruker 150/600 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);ESI-TOF型高分辨质谱仪;NIKON A1型快速扫描成像共聚焦显微镜;MC170HD型倒置显微镜。
Percoll液、1X磷酸盐缓冲液、二甲基亚砜。其余所用试剂为分析纯或化学纯。
1.2 合成
(1) 化合物1的合成
向100.00 mL反应瓶中加入氨基甲酸乙酯2.67 g(30.00 mmol),无水二氯甲烷30.00 mL,冰浴搅拌15 min。加入6.00 mL(75.00 mmol)草酰氯,并在冰浴下反应1 h。撤去冰浴,于70 ℃下加热回流4 h,冷却至室温后,减压浓缩除去溶剂,得到白色黏稠液体化合物1,不经纯化直接用于下一步反应。
(2) 化合物2的合成(以2a为例)
向100.00 mL反应瓶中加入苄胺3.21 g(30.00 mmol),三乙胺3.03 g(30.00 mmol),二氯甲烷30.00 mL。冰浴搅拌15 min后,缓慢加入二硫化碳7.61 g(100.00 mmol),之后迅速加入EDCI(缩合剂)5.76 g(30.00 mmol),冰浴条件下反应30 min。撤去冰浴,于室温反应16 h,TLC监测(乙酸乙酯/石油醚=5/95,V/V)反应基本完全。减压浓缩除去溶剂,加入乙酸乙酯100.00 mL进行稀释,加入饱和碳酸氢钠溶液(3×20.00 mL)和饱和食盐水(2×20.00 mL)洗涤,有机相用无水硫酸钠干燥,旋转蒸发除去溶剂。残余物经硅胶(100~200目)柱层析(洗脱剂:乙酸乙酯/石油醚=2/98,V/V),分离得到3.58 g(24.00 mmol)无色透明液体2a,产率80.30%; 4.01 g无色透明液体2b,产率80.00%。
(3) 化合物3的合成(以3a为例)
向100.00 mL反应瓶中加入化合物12.30 g(20.00 mmol),化合物2a苄基硫代异氰酸酯2.98 g(20.00 mmol),正己烷12.00 mL和二氯甲烷12.00 mL。冰浴下搅拌15 min后,加入磺酰氯2.70 g(20.00 mmol),冰浴下反应30 min。撤去冰浴,室温反应16 h后移除干燥管,与空气密切接触30 min,TLC监测(乙酸乙酯/石油醚=1/9,V/V)反应原料基本消耗完全。减压浓缩除去溶剂,加入乙酸乙酯100.00 mL进行稀释,加入饱和碳酸氢钠溶液,调节pH值为中性,采用饱和食盐水(2×20.00 mL)洗涤,有机相用无水硫酸钠干燥,旋转蒸发除去溶剂。残余物经硅胶(100~200目)柱层析(洗脱剂:乙酸乙酯/石油醚=1/9,V/V),乙醇重结晶纯化得到化合物3a。
3a:白色粉末3.60 g,产率64.30%;1H NMR(600 MHz, CDCl3)δ: 7.45(d,J=7.0 Hz, 2H), 7.37~7.30(m, 3H), 4.84(s, 2H), 4.41(q,J=7.1 Hz, 2H), 1.39(t,J=7.0 Hz, 3H);13C NMR(151 MHz, CDCl3)δ: 164.13, 148.30, 148.23, 134.36, 129.21, 128.84, 128.62, 65.50, 45.95, 14.21。
3b:白色粉末3.66g,产率61.40%;1H NMR(600 MHz, CDCl3)δ: 7.44(dd,J=8.6 Hz, 5.3 Hz, 2H), 7.01(t,J=8.6 Hz, 2H), 4.80(s, 2H), 4.41(q,J=7.1 Hz, 2H), 1.39(t,J=7.1 Hz, 3H);13C NMR(151 MHz, CDCl3)δ: 164.12, 162.84(d,JC-F=247.8 Hz), 148.21(d,JC-F=16.1 Hz), 131.27(d,JC-F=8.4 Hz), 130.23(d,JC-F=3.3 Hz), 115.84, 115.70, 65.56, 45.17, 14.20;19F NMR(565 MHz, CDCl3)δ: -112.96。
(4) 化合物4的合成(以4a为例)
向50.00 mL反应瓶中加入化合物3a固体粉末1.00 g(3.50 mmol),二氯甲烷10.00 mL,待完全溶解后加入乙二胺1.00 mL,室温下反应3 h。TLC监测(乙酸乙酯/石油醚=3/7,V/V)原料基本消耗完全。冰浴条件下,加入一定量的甲酸中和乙二胺(EDA),调节pH为中性,减压浓缩除去溶剂。加入乙酸乙酯50.00 mL稀释,加入水(2×15.00 mL)和饱和食盐水(2×15.00 mL)洗涤,有机相用无水硫酸钠干燥,旋转蒸发除去溶剂。残余物经硅胶(100~200目)柱层析(洗脱剂:乙酸乙酯/石油醚=1/9~2/8,V/V),纯化得到化合物4a。
4a:白色固体粉末0.67 g,产率91.40%;1H NMR(600 MHz, CDCl3)δ: 7.41(d,J=7.3 Hz, 2H), 7.38~7.30(m, 3H), 4.83(s, 2H);13C NMR(151 MHz, CDCl3)δ: 167.92, 155.55, 134.86, 128.82, 128.67, 128.42, 45.56。
4b:白色固体粉末0.60 g,产率80.10%;1H NMR(600 MHz, CDCl3)δ: 7.41(dd,J=8.5, 5.4 Hz, 2H), 7.02(t,J=8.6 Hz, 2H), 4.79(s, 2H);13C NMR(151 MHz, CDCl3)δ: 167.72, 162.72(d,JC-F=247.4 Hz), 155.36, 130.81(d,JC-F=8.3 Hz), 130.71(d,JC-F=3.3 Hz), 115.73(d,JC-F=21.6 Hz), 44.86;19F NMR(565 MHz, CDCl3)δ: -113.34。
(5) 化合物5的合成
向100.00 mL反应瓶中加入1,8-萘二甲酸酐1.98 g(10.00 mmol), 4-氨基-1-丁醇1.78 g(20.00 mmol),无水乙醇40.00 mL,于90 ℃下加热回流6 h。TLC监测(乙酸乙酯/石油醚=3/7,V/V)反应完全后,停止加热,冷却至室温。将溶液倒入大量冰水中,有白色固体析出,抽滤,烘干,得到2.41 g(8.96 mmol)白色固体5,粗产率89.60%,不经纯化处理直接用于下一步反应。
(6) 化合物6的合成
向50.00 mL反应瓶中加入化合物50.54 g(2.00 mmol),四溴化碳1.06 g(3.20 mmol),二氯甲烷15.00 mL,冰浴下搅拌15 min。随后加入三苯基膦0.84 g(3.20 mmol),反应30 min后撤去冰浴,室温反应16 h,TLC监测(乙酸乙酯/石油醚=2/8,V/V)反应原料消耗完全。减压浓缩除去溶剂,残余物经硅胶柱(100~200目)层析(洗脱剂:乙酸乙酯/石油醚=15/85,V/V),纯化得到白色固体粉末化合物60.53g,产率80.30%;1H NMR(600 MHz, CDCl3)δ: 8.53(d,J=8.0 Hz, 2H), 8.16(d,J=7.7 Hz, 2H), 7.70(t,J=7.5 Hz, 2H), 4.18(t,J=7.2 Hz, 2H), 3.45(t,J=6.7 Hz, 2H), 2.00~1.93(m, 2H), 1.91~1.85(m, 2H);13C NMR(151 MHz, CDCl3)δ: 164.09, 133.94, 131.50, 131.21, 128.04, 126.91, 122.49, 39.33, 33.21, 30.26, 26.89。
(7) 化合物7的合成
向100.00 mL反应瓶中加入4-溴-1,8-萘酐2.77 g(10.00 mmol), 4-氨基-1-丁醇1.33 g(15.00 mmol),无水乙醇30.00 mL,并于90 ℃下加热回流6 h。TLC监测(乙酸乙酯/石油醚=3/7,V/V)反应完全。静置冷却至室温,将溶液倒入大量冰水中,有固体析出,抽滤,烘干,得到2.68 g(7.70 mmol)淡黄色固体7,粗产率77.00%,不经纯化处理直接用于下一步反应。
(8) 化合物8的合成
向100.00 mL反应瓶中加入化合物72.68 g(7.7 mmol),吗啉2.01 g(23.10 mmol),乙二醇单甲醚30.00 mL,于125 ℃下加热回流6 h。TLC监测(乙酸乙酯/石油醚=4/6,V/V)反应完全。静置冷却至室温,将溶液倒入大量冰水中,有固体析出,抽滤,烘干,得到黄色固体。粗产物经硅胶(100~200目)柱层析(洗脱剂:乙酸乙酯/石油醚=3/7~5/5,V/V),纯化得到黄色固体粉末化合物81.82g,产率66.80%;1H NMR(600 MHz, CDCl3)δ: 8.56(d,J=7.1 Hz, 1H), 8.51(d,J=8.0 Hz, 1H), 8.40(d,J=8.3 Hz, 1H), 7.69(t,J=7.8 Hz, 1H), 7.21(d,J=8.0 Hz, 1H), 4.20(t,J=7.8 Hz, 2H), 4.01(t,J=7.0 Hz, 4H), 3.72(t,J=6.4 Hz, 2H), 3.26(t,J=3.8 Hz, 4H), 1.87~1.78(m, 2H), 1.73~1.65(m, 2H);13C NMR(151 MHz, CDCl3)δ: 164.48, 164.03, 155.67, 132.60, 131.23, 130.12, 129.86, 126.11, 125.86, 123.24, 117.07, 114.97, 66.97, 62.48, 53.44, 39.78, 29.91, 24.52。
(9) 化合物9的合成
向50.00 mL反应瓶中加入化合物80.71 g(2.00 mmol),乙酸乙酯20.00 mL,冰浴下搅拌15 min。加入三溴化磷0.50 mL(5.50 mmol),冰浴下反应30 min。撤去冰浴,于90 ℃下加热回流2 h,TLC监测(乙酸乙酯/石油醚=3/7,V/V)反应原料消耗完全,静置冷却至室温。加入适量的甲醇中和多余的三溴化磷,直至不再冒烟为止。加入30.00 mL乙酸乙酯进行稀释,再加入饱和碳酸氢钠溶液,调节pH为中性,用饱和食盐水(2×10.00 mL)洗涤,有机相用无水硫酸钠干燥,旋转蒸发除去溶剂。残余物经硅胶(100~200目)柱层析(洗脱剂:乙酸乙酯/石油醚=2/8~3/7,V/V),纯化得到黄色固体粉末化合物90.53 g,产率63.90%;1H NMR(600 MHz, CDCl3)δ: 8.57(d,J=8.0 Hz, 1H), 8.52(d,J=8.0 Hz, 1H), 8.42(d,J=9.1 Hz, 1H), 7.70(t, 8.2 Hz, 1H), 7.22(d,J=8.0 Hz, 1H), 4.20(t,J=7.2 Hz, 2H), 4.02(t,J=6.9Hz, 4H), 3.46(t,J=6.7 Hz, 2H), 3.26(t,J=6.5 Hz, 4H), 2.00~1.94(m, 2H), 1.92~1.86(m, 2H);13C NMR(151 MHz, CDCl3)δ: 164.40, 163.93, 155.68, 132.60, 131.24, 130.14, 129.87, 126.12, 125.86, 123.20, 117.02, 114.97, 66.97, 53.45, 39.21, 33.24, 30.27, 26.92。
(10) 目标化合物S的合成(以S1为例)
向50.00 mL反应瓶中加入化合物60.33 g(1 mmol),化合物4a0.21 g(1.00 mmol),无水乙腈10.00 mL。室温搅拌15 min后,加入碳酸钾0.28 g(2.00 mmol),室温反应24 h。TLC监测(乙酸乙酯/石油醚=3/7,V/V)反应原料基本消耗完全,停止反应,减压浓缩除去溶剂。加入乙酸乙酯50.00 mL稀释,水(2×10.00 mL)和饱和食盐水(2×10.00 mL)洗涤,有机相用无水硫酸钠干燥,旋转蒸发除去溶剂。残余物经硅胶(100~200目)柱层析(洗脱剂:乙酸乙酯/石油醚=1/9~2/8,V/V),乙醇重结晶,纯化得到目标化合物S1。
S1:白色固体粉末0.21 g,产率45.60%;1H NMR(600 MHz, CDCl3)δ: 8.59(d,J=7.2 Hz, 2H, 23, 29-H), 8.22(d,J=8.1 Hz, 2H, 25, 27-H), 7.76(t,J=7.7 Hz, 2H, 24, 28-H), 7.42(d,J=7.1 Hz, 2H, 8, 12-H), 7.34~7.27(m, 3H, 9, 10, 11-H), 4.80(s, 2H, 6-H), 4.22(t,J=7.0 Hz, 2H, 16-H), 3.71(t,J=7.0 Hz, 2H, 13-H), 1.84~1.78(m, 2H, 15-H), 1.78~1.73(m, 2H, 14-H);13C NMR(151 MHz, CDCl3)δ: 165.98(C5), 164.23(C18,22), 152.95(C3), 135.24(C25,27), 134.08(C23,29), 131.61(C7), 131.36(C24,28), 128.87(C9,11), 128.70(C26), 128.21(C8,12), 128.18(C10), 126.99(C19,21), 122.54(C20), 45.97(C13), 44.64(C6), 39.42(C16), 26.20(C15), 25.03(C14); HR-MS(ESI)m/z: calcd for C25H21N3O4NaS 482.1147{[M+Na]+} found 482.1145。
S2:白色固体粉末0.18 g,产率37.50%;1H NMR(600 MHz, CDCl3)δ: 8.58(d,J=7.3 Hz, 2H, 23, 29-H), 8.22(d,J=8.2 Hz, 2H, 25, 27-H), 7.76(t,J=7.7 Hz, 2H, 24, 28-H), 7.41(dd,J=8.4 Hz, 5.4 Hz, 2H, 8, 12-H), 6.98(t,J=8.6 Hz, 2H, 9, 11-H), 4.76(s, 2H, 6-H), 4.21(t,J=7.0 Hz, 2H, 16-H), 3.70(t,J=6.9 Hz, 2H, 13-H), 1.83~1.78(m, 2H, 15-H), 1.78~1.72(m, 2H, 14-H);13C NMR(151 MHz, CDCl3)δ: 165.93(C5), 164.21(C18,22), 162.62(C10)(d,JC-F=246.9 Hz), 152.83(C3), 134.08(C25,27), 131.60(C23,29), 131.33(C24,28), 131.11(C7)(d,JC-F=3.2 Hz), 130.88(C8,12)(d,JC-F=8.3 Hz), 128.15(C26), 126.98(C19,21), 122.50(C20), 115.59(C9,11)(d,JC-F=21.5 Hz), 45.21(C13), 44.64(C6), 39.41(C16), 26.20(C15), 25.02(C14);19F NMR(565 MHz, CDCl3)δ: -113.65; HR-MS(ESI)m/z: calcd for C25H20FO4NaS 500.1050{[M+Na]+} found 500.1050。
S3:黄绿色油状液滴0.16g,产率29.60%;1H NMR(600 MHz, CDCl3)δ: 8.58(d,J=6.5 Hz, 1H, 27-H), 8.52(d,J=8.0 Hz, 1H, 29-H), 8.43(d,J=8.9 Hz, 1H, 23-H), 7.71(t,J=8.0 Hz, 1H, 28-H), 7.42(d,J=7.2 Hz, 2H, 8,12-H), 7.33~7.27(m, 3H, 9, 10, 11-H), 7.23(d,J=8.1 Hz, 1H, 24-H), 4.80(s, 2H, 6-H), 4.20(t,J=7.0 Hz, 2H, 16-H), 4.02(t,J=6.8 Hz, 4H, 32, 34-H), 3.70(t,J=7.1 Hz, 2H, 13-H), 3.27(t,J=7.2 Hz, 4H, 31, 35-H), 1.82~1.77(m, 2H, 15-H), 1.76~1.72(m, 2H, 14-H);13C NMR(151 MHz, CDCl3)δ: 166.01(C5), 164.44(C25), 163.98(C18), 155.77(C22), 152.93(C3), 135.24(C23), 132.68(C29), 131.31(C7), 130.22(C27), 129.90(C28), 128.85(C9,11), 128.70(C26), 128.20(C8,12), 126.14(C10), 125.88(C19), 123.17(C20), 116.96(C21), 115.00(C24), 66.97(C32,34), 53.45(C31,35), 45.96(C13), 44.66(C6), 39.26(C16), 26.18(C15), 25.05(C14); HR-MS(ESI)m/z: calcd for C29H28N4O5NaS 567.1662{[M+Na]+} found 567.1672。
1.3 目标化合物S1, S2, S3对精子的荧光标记测试
(1) 新鲜精液标本的采集
本实验所用样本来自男性志愿者,年龄20~35岁,禁欲3~7 d,手淫法获取精液。精液样本要求:液化时间≤30 min,精液量≥2.00 mL, pH=7.2~8.0,白细胞﹤1×106mL-1,精子密度≥15×106mL-1,精子的运动活力满足前向运动率(Progressive)>32%,精浆和血清抗精子抗体阴性。本研究所有精液样本均取得捐献者知情同意且所有标本在实验结束后均进行高温煮沸消杀。
(2) 高活力精子的获取
采用Percoll离心法收集高活力精子,1×PBS与Percoll原液1 ∶1混合后得到50%(V/V)的Percoll工作液。将Percoll工作液加入15.00 mL离心管中,在Percoll工作液上方加入等体积充分混匀的精液,于25 ℃条件下以500.00 g离心15 min,弃去精子沉淀团上大部分上清液,1×PBS重悬精子沉淀团,以300.00 g离心5 min洗涤精子,重复洗涤两次,用BWW培养基重悬精子团沉淀,并稀释调整精子浓度为10~20×106mL-1,备用。
(3) 目标化合物快速杀精制动活性的测定
取化合物S1,S2,S3用BWW进行倍比稀释,最高浓度为100 μM。将稀释后的化合物加入96孔板,每孔50 μL。取Percoll离心重悬后的高活力精子,用BWW稀释调整精子浓度为10~20×106mL-1,吸取50 μL精子悬液与96孔板中化合物混合的同时按下秒表计时20 s。以溶剂DMSO为阴性对照,Tideglusib为阳性对照,显微镜下观察该浓度下20 s内该化合物能否制动全部精子,由此测出全部精子丧失运动能力所需药物的最小有效浓度(MEC)。此实验至少重复3次,得MEC平均值。
(4) 目标化合物对精子的荧光标记测试
取Percoll离心重悬后的高活力精子,用BWW稀释调整精子浓度为10~20×106mL-1,并平均分为7组,分别命名为Tideglusib组、S1-5 μM组、S1-10 μM组、S2-5 μM组、S2-10 μM组、S3-5 μM组、S3-10 μM组。向各组中加入相应的化合物并调整至相应的浓度,反应20 s, 500 g离心5 min弃上清,1×PBS洗涤2次后重悬,涂片,室温下避光晾干,荧光封片剂封片后激光共聚焦显微镜下观察,此实验全程避光处理。
(5) 统计学分析
采用Graphpad prism9.0对数据进行统计学分析,MEC浓度结果以平均数±标准差(mean±SD)表示,组间比较采用Unpaired t test, P﹤0.05定为具有统计学差异。
2 结果与讨论
2.1 目标化合物的MEC测定
测得目标化合物与其先导化合物Tideglusib在20 s内快速制动100%精子所需的最小化合物浓度(MEC值)见表1。目标化合物与先导化合物Tideglusib相比,除S2外,无显著性差异。测试结果表明,荧光标记的衍生化合物与其先导化合物Tideglusib相同,仍具有很强的精子制动活性,可用于标示和定位研究Tideglusib与细胞作用的模式及其生物学靶标。

表1 目标化合物与Tideglusib在20 s内使人精子100%制动的最小浓度MEC值的比较/μMTable 1 Comparison of the minimum concentration MEC value of target compound and tideglusib for 100% immobilization of human sperm within 20 s/μM
2.2 目标化合物对人精子的荧光标记作用
为了验证目标化合物对人精子的标记作用,分别设置了5 μM、 10 μM两个不同浓度梯度的化合物对收集的高活力精子进行荧光染色。目标化合物对精子的典型染色结果见图1。结果显示,与没有荧光的Tideglusib相比,化合物S1,S2对精子具有明显的蓝色荧光标记作用,而化合物S3对精子有黄绿色荧光标记作用。S1,S2对精子的荧光染色部位主要集中在精子尾部颈段和中段位置及精子头部顶体后区位置。S3对精子的荧光染色部位主要集中在精子尾部颈段和中段(middle piece)位置。这些标记部位与电镜观察到的Tideglusib对精子的尾部颈段和中段位置以及精子头部质膜的损伤部位基本一致[11],表明荧光标记的衍生化合物,保持了先导化合物Tideglusib的活性,后续可用于Tideglusib与细胞作用的模式及其生物学靶标和活性机制的研究。

图1 目标化合物S1、 S2、 S3及先导化合物Tideglusib对人精子的荧光染色Figure 1 Fluorescent staining of human sperm by target compounds S1, S2, S3 and lead compound Tideglusib
3 结论
本文合成了3个带有荧光基团的1,2,4-噻二唑-3,5-二酮化合物S1,S2,S3。目标化合物结构通过1H NMR、13C NMR、 HR-MS表征验证;测定了目标化合物对精子运动活性的抑制作用及其荧光标记在精子上的分布模式。结果表明:目标化合物S1,S2,S3保持了以先导化合物Tideglusib为代表的噻二唑二酮类化合物快速杀精制动活性;目标化合物对精子荧光染色主要集中在精子尾部颈段和中段位置及精子头部后区,与电镜观察到的Tideglusib导致精子尾部中段和精子头部质膜损伤的位置基本一致,进一步证明了噻二唑二酮类化合物可能是通过损伤精子尾部颈段和中段及精子头部来抑制精子运动。目标化合物对体细胞的染色很弱,这也与Tideglusib的精子选择性活性(低体细胞毒性)相一致。总之,目标化合物在引入荧光标记的同时,成功保存了先导化合物的生物学活性,为探究噻二唑二酮类化合物与靶细胞的作用模式,以及明确其活性的生物学靶标及分子机制提供了新的工具和思路,为开发研究新的医药中间体提供了方向。
