己二酰氯的绿色制备工艺
2022-08-31吴文静戎有明周建成李乃旭
吴文静 王 楠 戎有明 周建成, 李乃旭,*
(1. 东南大学 化学化工学院,江苏 南京 211189; 2. 中微纳米功能材料研究院有限公司,江苏 南京 210044)
酰氯作为最活泼的酰基化试剂,可以与氨/胺反应生成酰胺,与醇反应生成酯,与羧酸根离子反应生成酸酐等[1]。其中己二酰氯作为有机中间体有多种用途,如己二酰氯与金刚石薄膜表面的官能团结合可得共价吸附层[2];已二胺和二酰氯之间进行界面缩聚是一种生产尼龙66的新方法[3];对称二价分子被研究用于治疗多种疾病[4],例如将己二酰氯参与反应合成的负载替莫唑胺(TMZ)的磁铁矿三嵌段共聚物用于药物递送[5];近年来用己二酰氯制备的支化大分子或偶联剂将典型的一维碳纳米管结合到碳纤维表面形成“柔性—刚性”多尺度增强结构,是一种提高碳纤维增强复合材料界面性能的有效方法[6];己二酰氯与N,N′-己烷-1,6-二基-双苯甲酰胺作用能够产生可通过血脑屏障的毒蕈碱受体,这种受体可用于影响激动剂和拮抗剂与受体蛋白正构位点的结合[7];相比较于直接酯化的苛刻反应条件,使用己二酰氯可在相转移催化条件下对单或双取代酚进行酯化得到单酯或二酯,且获得了更高的产物收率[8]。
酰氯的传统制备方法中的亚硫酰氯法一般将N,N-二甲基甲酰胺(DMF)作催化剂与亚硫酰氯反应得到酰氯[9],亚硫酰氯沸点低,不易制备高沸点酰氯,且对设备腐蚀严重。三氯化磷法一般不需要结合催化剂[10],适合制备低沸点酰氯,但生成的亚磷酸易氧化成磷酸,具有腐蚀性,且受热分解产生毒性气体氧化磷。草酰氯法一般将其与DMF形成Vilsmeier中间体参与反应[11],原料为无色发烟液体,气味刺鼻,反应过程剧烈不易控制。氰尿酰氯法的原料具有明显刺激作用,破坏环境造成水体污染等[12,13]。这些方法均不符合绿色生产工艺标准。为解决酰氯化试剂毒性强、易挥发及溶剂回收困难等问题,本文采用三氯甲苯结合过渡金属氯化物制备己二酰氯[14-16],同时联产苯甲酰氯(如图1),该合成路线简单安全,为己二酰氯新工艺的开发以及应用提供了一定的借鉴价值。
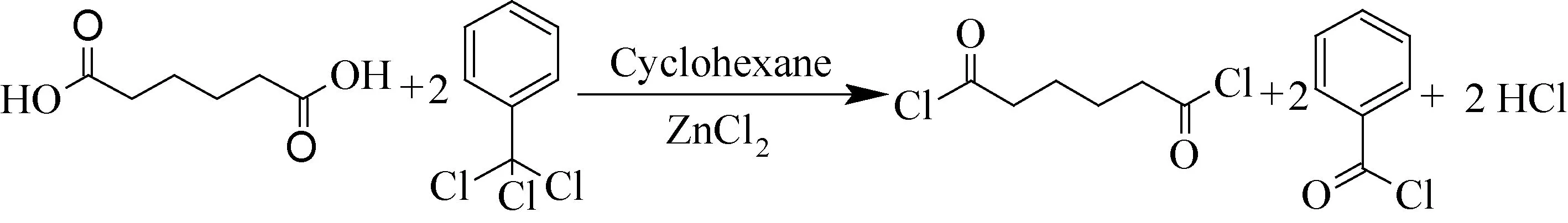
图1 己二酰氯的制备
1 实验部分
1.1 主要仪器与试剂
仪器:气相色谱仪GC-9860(上海奇阳信息科技有限公司),傅里叶红外光谱仪IR-408(日本岛津公司),600 MHz核磁共振波谱仪AVANCE III HD(美国Bruker公司)。
试剂:己二酸,AR,上海易恩化学技术有限公司;环己烷,AR,上海易恩化学技术有限公司;三氯甲苯,99%,国药集团化学试剂有限公司;四氢呋喃,≥99.8%,国药集团化学试剂有限公司;甲苯,≥99.5%,国药集团化学试剂有限公司;ZnCl2,AR,国药集团化学试剂有限公司;AlCl3,AR,上海易恩化学技术有限公司;FeCl3,AR,上海易恩化学技术有限公司。
1.2 实验方法
将一定量的己二酸、环己烷与三氯甲苯加入三口烧瓶中,搅拌,再称取少量ZnCl2加入反应体系,30 ℃下反应3 h; 反应结束后于-0.05 MPa下旋蒸除去环己烷,减压蒸馏后得到淡黄色澄清液体为己二酰氯。
1.3 结果与表征
己二酰氯的核磁氢谱见图2,化学位移值δ1.57,1.25,0.88 ppm的小峰,峰高不大于1个质子,为杂质峰,其余2组主要峰的积分比为1∶1,对应两个4重峰的图谱中有2种质子。总积分值扣除杂质峰按8个质子分配,化学位移值δ2.94 ppm为邻近羰基旁1、2号位H的信号峰,由于与羰基相连,p-π共轭,亚甲基氢化学位移值移向低场;化学位移值δ1.78 ppm为中间两种3、4号位H的信号峰。1H NMR (600 MHz, CDCl3) 2.94 (tq, J=1.44,1.4 Hz), 1.97-1.17 (m)。
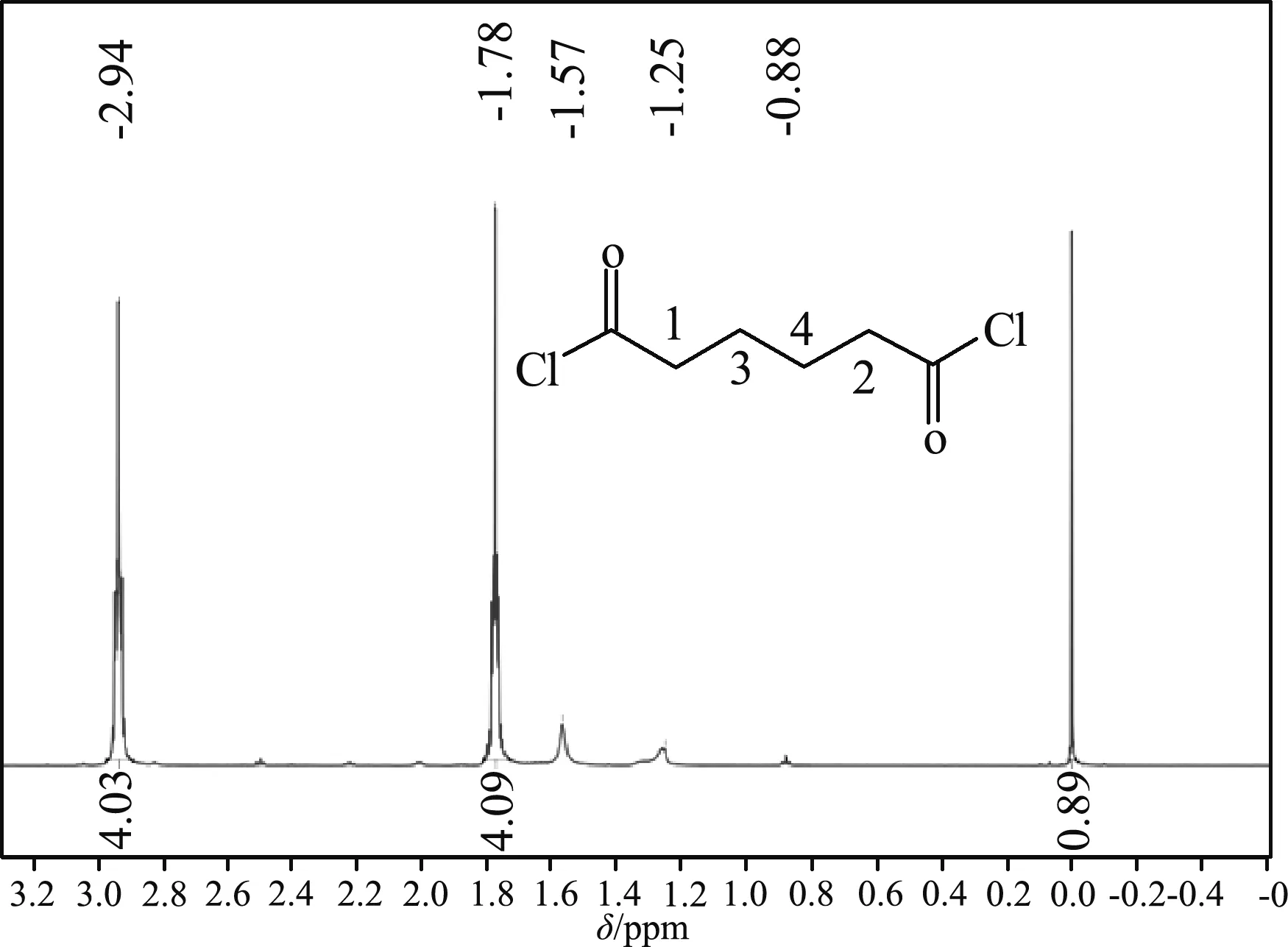
图2 己二酰氯的核磁氢谱
己二酰氯的核磁碳谱见图3,化学位移值δ173.10 ppm为连接杂原子的羰基中1、2号位C的信号峰,化学位移值δ23.57 ppm为不直接连接杂原子的脂肪链碳原子即3、4、5、6号位C的信号峰,δ46.21 ppm为C-Cl即同样是1、2号位C的信号峰。
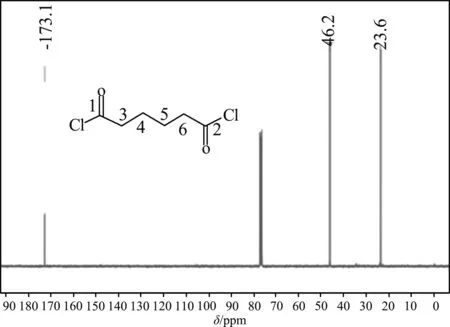
图3 己二酰氯的核磁碳谱
己二酰氯的红外谱图见图4,由图4分析结构式的不饱和度为2,表明分子中有两个C=C或C=O,3 300~2 800 cm-1区域为C-H伸缩振动,低于且接近3 000 cm-1的2 950 cm-1、2 877 cm-1一般为饱和C-H的伸缩振动吸收,1 805 cm-1和1 693 cm-1的峰属于己二酰氯中脂肪酮的强伸缩振动吸收,1 407 cm-1的吸收峰表明己二酰氯中羰基伸缩振动峰,脂肪族C-Cl伸缩振动在850~550 cm-1区域有744 cm-1吸收峰,与产物的标准红外光谱图基本一致。

图4 己二酰氯的红外谱图
2 单因素实验
2.1 时间因素
在己二酸与三氯甲苯摩尔比为1∶2,27 mL环己烷,0.1 g ZnCl2,反应温度30 ℃的条件下设置反应时间为3~7 h,己二酰氯收率与时间的关系如图5。
由图5可以看出,具有强亲电能力的Lewis酸ZnCl2发生亲电取代反应,前5 h收率逐渐上升,反应液中酸根离子与苄基正离子浓度逐渐增大,反应时间为5 h达最大值51.60%。5 h前消耗的酸根离子与己二酸产生配合物,但时间过短反应远未达终点,体系中可能还有大量酸根离子及苄基正离子未参与到与己二酸形成中间体的步骤中。5 h后,体系副反应发生的概率增大。综上,最适宜的反应时间为5 h。
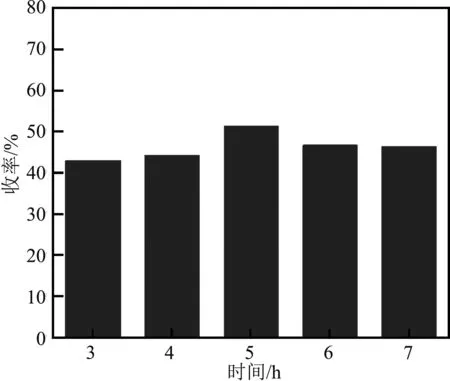
图5 时间对收率的影响
2.2 温度因素
己二酸与三氯甲苯摩尔比为1∶2,27 mL环己烷,0.1 g ZnCl2,设置反应温度40 ℃~80 ℃,反应5 h。己二酰氯收率与温度的关系如图6。
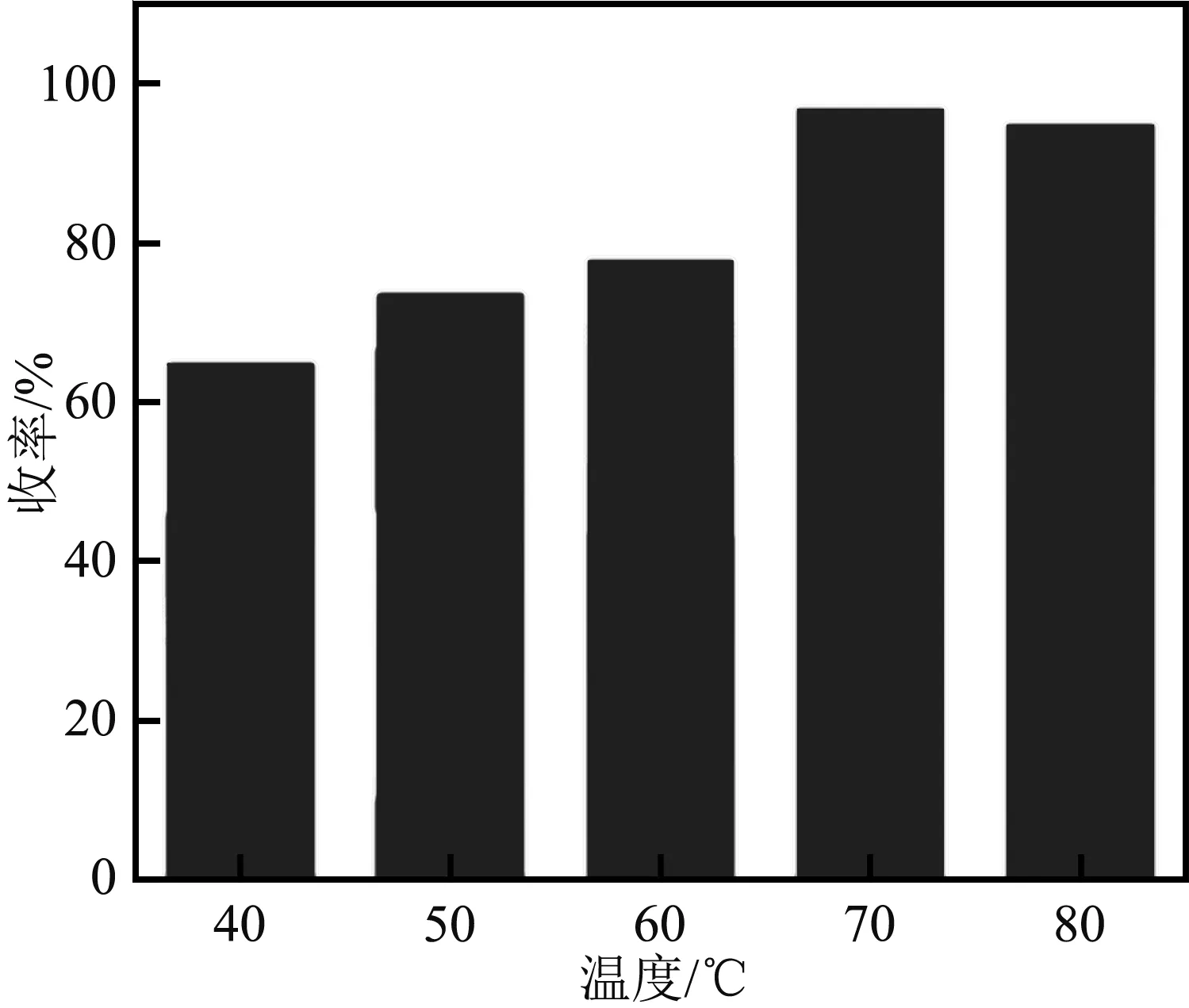
图6 温度对收率的影响
由图6可知,40 ℃~70 ℃范围内反应收率逐渐上升,在70 ℃达最大值96.88%,随后下降。温度作为宏观量,高温时分子热运动剧烈导致反应速率增大,低温时反应速率慢,同样的反应时间内反应效率低,温度由60 ℃到70 ℃时的收率提高幅度最大。温度越高,内能越大,但同时伴随溶剂汽化挥发,温度过高,反应效果差。综上,最适宜的反应温度为70 ℃。
2.3 催化剂因素
己二酸与三氯甲苯摩尔比为1∶2,27 mL环己烷,0.1 g ZnCl2,70 ℃下反应5 h。选择三种Lewis酸催化剂,分别是:分子晶体AlCl3、FeCl3及离子晶体ZnCl2,己二酰氯收率与催化剂种类的关系如图7。
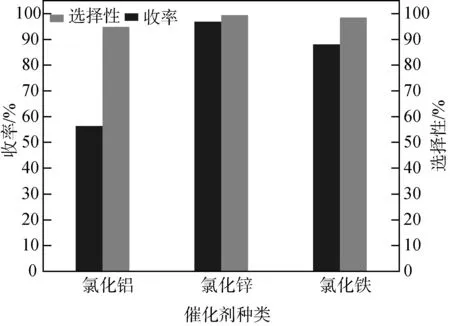
图7 催化剂对收率的影响
实际使用过程中,FeCl3与AlCl3在常温下具有强烈的吸水性,迅速吸收空气中的水分并潮解,放出HCl形成酸雾,而酰氯需要在严格无水的环境下制备,所以这两种催化剂制备条件相较具有强正负离子间静电作用力的ZnCl2更加苛刻,三种催化剂使用过程中发现ZnCl2吸水性最弱,反应的加料量更易于控制。从图7看出ZnCl2作催化剂时,收率最高。酸根离子与己二酸形成的配合物能够被苄基正离子准确进攻且大部分用于活化配合物中的氧,其他位置几乎不反应,反应完成后能够分解重新还原成Lewis酸催化剂循环参与反应。综上,最佳催化剂为ZnCl2。
2.4 溶剂因素
己二酸与三氯甲苯摩尔比为1∶2,0.1 g ZnCl2,70 ℃下反应5 h。选择三种溶剂,分别是:甲苯、环己烷、四氢呋喃。己二酰氯收率与溶剂种类关系如图8。
由于产物己二酰氯的极性弱,对比三种溶剂的极性大小,四氢呋喃极性最强,甲苯次之,环己烷极性最弱,根据相似相溶原理,环己烷与产物的极性差异度最小,从图8明显看出环己烷作为溶剂时,产物的收率最高,原料选择性高。综上,最佳溶剂为环己烷。
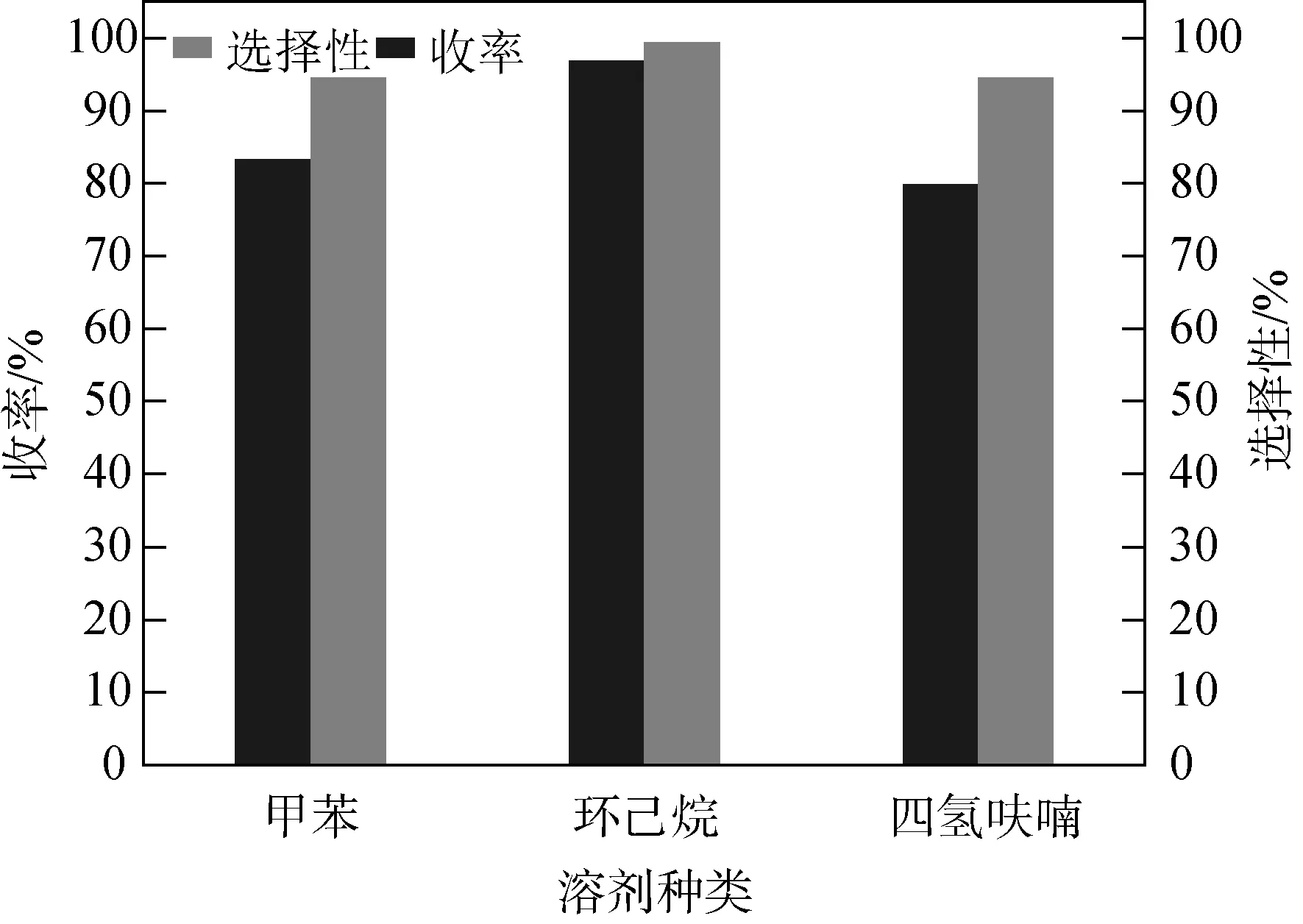
图8 溶剂对收率的影响
3 反应机理
由于Lewis酸具有空轨道,吸电子能力强,在Lewis酸活化下,三氯甲苯中的氯易于离去,分别形成带负电荷的酸根离子和带正电荷的苄基离子;酸根离子活化己二酸中的活泼氢使其离去,二者再结合形成配合物;带正电荷的苄基离子对配合物中的氧进行活化,使C-O键断裂,形成C-Cl键,进而分解还原得到己二酰氯、苯甲酰氯和Lewis酸。反应机理如图9。

图9 反应机理
4 结论
该路线以己二酸与三氯甲苯为起始底物,合成己二酰氯的同时联产苯甲酰氯,经过单因素实验确定时间为5 h,温度为70 ℃,催化剂为ZnCl2以及溶剂为环己烷,最终以96.88%的收率得到目标产物己二酰氯。对目标产物进行核磁与红外结构表征,均证实产物的基本结构。本文提出了一种制备己二酰氯的绿色合成新工艺,有效解决了使用传统酰氯化试剂存在的毒性强、易挥发及溶剂回收困难等问题,且具有进一步扩大生产与开发应用的价值。
