CDK2/4/6抑制剂PF-06873600的合成工艺研究
2022-08-31姜虹羽焦小雨邵俊兰唐春雷
丁 蕾 袁 昕 姜虹羽 焦小雨 邵俊兰 唐春雷
(江南大学 生命科学与健康工程学院,江苏 无锡 214122)
PF-06873600的化学名称为6-(二氟甲基)-8-((1R,2R)-2-羟基-2-甲基环戊基)-2-((1-(甲磺酰基)哌啶-4-基)氨基)吡啶基[2,3-d]嘧啶-7(8H)-酮,是由美国辉瑞制药公司研发的一种具有口服活性的、选择性的细胞周期蛋白依赖性激酶(CDKs)抑制剂[1]。CDKs属于丝氨酸/苏氨酸蛋白激酶家族,其激酶活性需要与细胞周期蛋白结合来激活,在细胞周期调节和转录过程中起着重要作用,通过靶向CDKs阻滞细胞增殖和分化已成为抗肿瘤的重要方法[2,3]。由辉瑞制药公司研发的CDK4/6抑制剂帕博西尼(Palbociclib),已经被FDA批准用于治疗乳腺癌[4],然而随着临床研究的不断发展,帕博西尼的临床疗效逐渐表现出耐药性[5]。Freeman-Cook, Kevin等人通过临床前模型和临床转录组标本的分析发现CDK2信号通路的激活作为一种补偿性机制导致了其耐药性[1]。基于帕博西尼的支架设计得到的PF-06873600在临床前研究中表现出较高的选择性和抗肿瘤活性,其对CDK2、CDK4和CDK6的酶抑制常数(Ki)分别为0.09 nM、0.13 nM和0.16 nM。目前,一项名为PF-06873600针对癌症患者的研究正处于临床Ⅱ期(NCT03519178)。PF-06873600有望在因对帕博西尼产生耐药性而复发的疾病中表现出有效的治疗活性。本文对PF-06873600的合成方法进行研究,以期为该化合物的合成以及结构类似物的研究提供理论参考。
目前,只有原研公司报道了PF-06873600的合成路线(图1)[6,7]:以1-甲基-6-氧杂双环[3,1,0]己烷(1)为起始原料,在100 ℃条件下以水为溶剂,与苄胺反应,反应液经萃取、旋蒸和正庚烷打浆后得到淡黄色固体产物2;随后用手性氨基酸进行手性拆分得到3;产物3在Pd(OH)2/C和H2条件下脱苄胺得到中间体4;以(4-氯-2-(甲硫基)嘧啶-5-基)甲醇为原料,异丙醇作为溶剂,在N,N-二异丙基乙胺催化下,与中间体4发生取代反应,得到产物5;随后产物5经二氧化锰氧化得到氧化产物6;产物6与乙酸乙酯在LiHMDS的催化下环合得到环合产物7;环合产物7再经氧化剂oxone氧化得到中间体8;之后1-甲砜基-4-氨基哌啶与中间体8发生取代反应得到产物9;随后以乙腈为溶剂,产物9与N-碘代琥珀酰亚胺发生取代反应得到碘代产物10;最后碘代产物10与(二氟甲基)三甲基硅烷反应得到终产物PF-06873600。
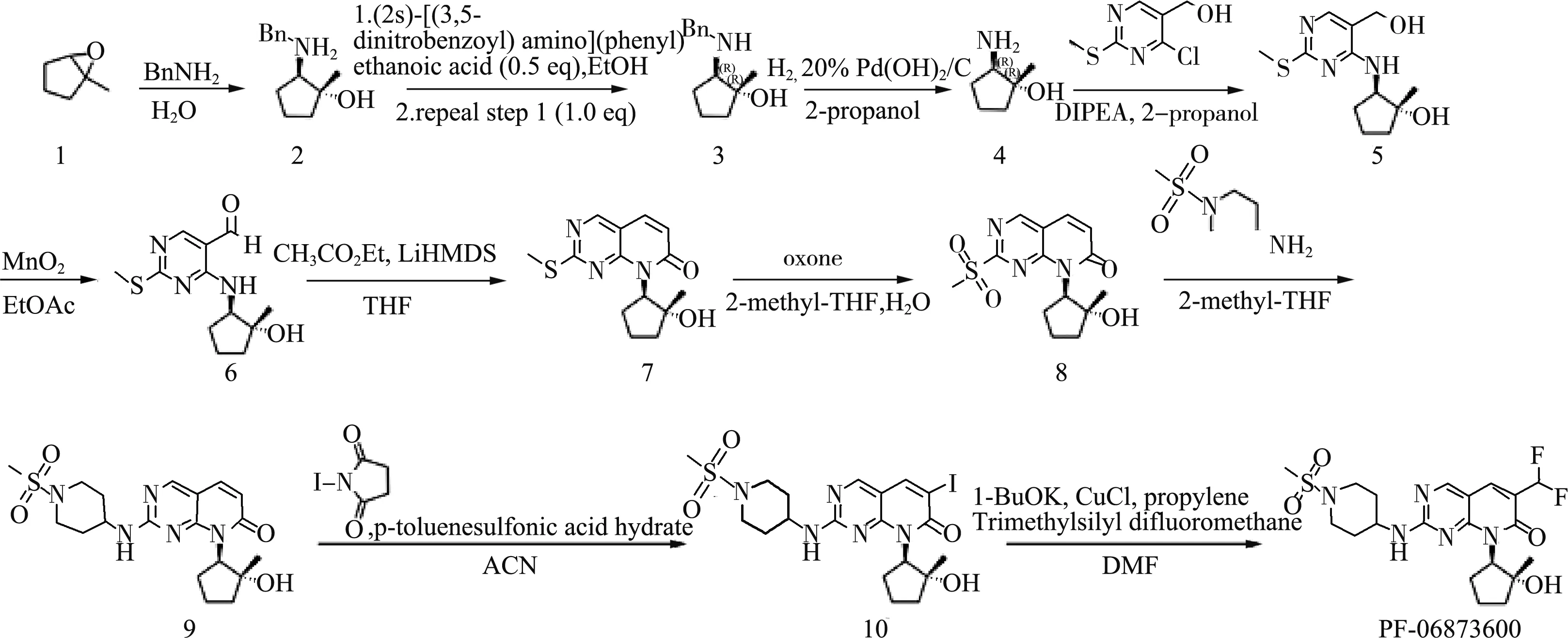
图1 PF-06873600的合成路线1
原研专利的合成路线步骤为10步反应,其合成起始原料较为昂贵,增加了工艺成本,另外部分中间体的合成过程较为复杂,限制了该合成路线在工业生产中的应用。本文以廉价易得的1-甲基环戊烯(K-1)为起始原料,经一步反应即可得到昂贵的1-甲基-6-氧杂双环[3,1,0]己烷(1),且收率高达95%,大大降低了工艺成本。本工艺路线中用更廉价的5-溴-2,4-二氯嘧啶替换(4-氯-2-(甲硫基)嘧啶-5-基)甲醇,经三步反应即可得到中间体9(K-6),较原研路线缩短了一步,且收率由原来的34.9%提升至36.2%。另外,在K-6的后处理中,经实验探索,采用正庚烷∶乙酸乙酯=10∶1作为打浆溶剂,节约了时间和溶剂成本。优化后的工艺路线在保证总收率的前提下,降低了生产成本和能耗,反应条件相对温和,后处理操作简便,适合工业化制备。合成路线见图2。

图2 PF-06873600的合成路线2
1 实验部分
1.1 主要仪器与试剂
LCMS-80质谱仪(日本岛津公司);LC1260色谱仪(安捷伦科技有限公司);Bruker AVII-400 MHZ 核磁共振仪(德国Bruker公司,TMS为内标);紫外线分析仪(郑州科泰实验设备有限公司)。实验所用试剂均为市售分析纯或化学纯,未进一步纯化处理。
1.2 合成步骤
1.2.1 2-(苄基氨基)-1-甲基环戊烷-1-醇(K-2)的制备
1-甲基环戊烯(30 g,365 mmol)溶于300 mL DCM中,冰浴条件下,慢慢滴加m-CPBA(94.53 g,547.5 mmol)的DCM(100 mL)混悬液,滴毕,移至室温,搅拌反应过夜。TLC监测原料反应完毕。反应液硅藻土过滤,滤液依次用10%硫代硫酸钠水溶液洗两遍、饱和碳酸氢钠水溶液洗三遍、水洗一遍,之后加入无水硫酸钠干燥,抽滤,滤液减压浓缩,得到淡黄色油状液体(34 g,收率95%),不做进一步纯化直接投入下一步反应。在装有100 mL水和苄胺(36.5 g,341 mmol)的反应瓶中加入淡黄色油状液体(33.5 g,341 mmol),将反应瓶移至100 ℃油浴中搅拌18 h,反应体系冷却至室温,移至冰浴中。反应液中先加适量水,然后用浓HCl调pH至1,乙酸乙酯萃取两遍,合并水相,并用NaOH调pH至10;再用乙酸乙酯萃取三遍,合并有机相,有机相中加无水硫酸钠,抽滤,滤液减压浓缩除去大部分溶剂得棕褐色油状液体。棕褐色油状液体在更高真空度的条件下浓缩除去一部分苄胺,然后向残留液体中加入正庚烷搅拌30 min,有淡黄色固体析出,抽滤,真空干燥后得到淡黄色固体49 g,收率为65.4%。MS-ESI, m/z:206[M+H]+。1H NMR (400 MHz, CDCl3)δ:7.38-7.32 (m, 4H), 7.28-7.23 (m, 1H), 3.91(d,J=13.2 Hz,1H), 3.80(d,J=13.3 Hz, 1H), 2.89(dd,J=8.7, 7.5 Hz, 1H), 2.12-2.07 (m, 1H), 1.80-1.59 (m, 5H), 1.37-1.31 (m, 1H), 1.26(s, 3H)。
1.2.2 (1R,2R)-2-(苄基氨基)-1-甲基环戊烷-1-醇((1R,1R)K-2)的制备
在单口瓶A中,将K-2(48 g,233.8 mmol)溶于EtOH(320 mL),加热至80 ℃,回流搅拌30 min。同时在配有内插温度计和冷凝管的三颈瓶B中,将手性氨基酸(2S)-[(3,5-二硝基苯甲酰基)氨基](苯基)乙酸(40.4 g,116.9 mmol,0.5eq)溶于EtOH(640 mL),加热至80 ℃,回流搅拌30 min,至白色固体溶解,溶液澄清。然后在80 ℃条件下将A瓶中的反应液慢慢加入B瓶中,可以观察到逐渐有固体产生,继续在80 ℃条件下搅拌4 h,反应瓶降温至室温,抽滤干燥后得白色固体46.7 g。将白色固体溶于水(200 mL)和乙酸乙酯(300 mL)的混合溶液中,向混合溶液中加入稀盐酸(4 M, 50 mL)搅拌30 s,可观察到有清晰的分层。分液得到水相和有机相。有机相进一步用稀盐酸(0.2 M, 25 mL×2)洗两遍,然后合并三次水相,在冰浴条件下用NaOH缓慢地将pH调至10,加入70 mL饱和NaCl稀释,用乙酸乙酯萃取4遍,合并有机相。有机相中加无水硫酸钠干燥,抽滤,滤液减压浓缩得白色固体(1R,2R)-2-(苄基氨基)-1-甲基环戊烷-1-醇(22.8 g),收率为95%。随后用1.0 eq的(2S)-[(3,5-二硝基苯甲酰基)氨基](苯基)乙酸将所得产物重复上述操作一次,最终得到白色固体22.1 g,收率为92%。1H NMR (400 MHz, CDCl3)δ:7.38-7.32 (m, 4H), 7.28-7.23 (m, 1H), 3.91 (d,J=13.2 Hz, 1H), 3.80(d,J=13.3 Hz, 1H), 2.89 (dd,J=8.7, 7.5 Hz, 1H), 2.12-2.07 (m, 1H), 1.80-1.59 (m, 5H), 1.37-1.31 (m, 1H), 1.26 (s, 3H)。
1.2.3 (1R,2R)-2-((5-溴-2-氯嘧啶-4-基)氨基)-1-甲基环戊烷-1-醇(K-3)的制备
将(1R,1R)K-2(22 g,107.2 mmol)溶于异丙醇(500 mL)中,加入Pd/C(2.2 g),于室温搅拌反应过夜,TLC监测反应完成。用硅藻土过滤反应液除去钯碳,滤饼用异丙醇洗两遍,滤液减压浓缩至溶剂剩余约300 mL,不做进一步纯化,直接用于下一步反应。在滤液中依次加入5-溴-2,4-二氯嘧啶(24.4 g,107.2 mmol)和三乙胺(10.7 g,107.2 mmol),反应液于室温下搅拌4 h,TLC监测反应完成。反应液减压浓缩后经柱层析得到白色固体25.2 g,收率为77%。MS-ESI, m/z:306,308[M+H]+。1H NMR (400 MHz, CDCl3)δ:8.17 (s, 1H), 5.47 (s, 1H), 4.27 (s, 1H), 4.24-4.18 (m, 1H), 2.30 (dt,J=8.3, 4.1 Hz, 1H), 2.00 (dd,J=11.4, 6.7 Hz, 1H), 1.88-1.73 (m, 3H), 1.57-1.50 (m, 1H), 1.15 (s, 3H)。
1.2.4 (1R,2R)-2-((5-溴-2-((1-(甲磺酰)哌啶-4-基)氨基)嘧啶-4-基)氨基)-1-甲基环戊烷-1-醇(K-4)的制备
将K-3(25 g,82 mmol)溶于DMSO中,依次加入N,N-二异丙基乙胺(12.7 g,98.4 mmol)和1-甲砜基-4-氨基哌啶(20.5 g,114.8 mmol),升温至100 ℃搅拌反应6 h,然后继续升温至110 ℃搅拌反应6 h,TLC监测反应完成。反应液用二氯甲烷稀释,水洗四遍,水相用二氯甲烷萃取,然后合并有机相。有机相中加入无水硫酸钠干燥,抽滤,滤液减压浓缩后经石油醚打浆得到白色固体24.9 g,收率为68%。MS-ESI, m/z:448,450[M+H]+。1H NMR (400 MHz, DMSO-d6)δ:7.86 (s, 1H), 6.83 (s, 1H), 5.78 (d,J=7.9 Hz, 1H), 4.61 (s, 1H), 4.24 (d,J=7.8 Hz, 1H), 3.74 (s, 1H), 3.53-3.48 (m, 2H), 2.86 (s, 3H), 2.81 (d,J=3.1 Hz, 2H), 1.92 (s, 2H), 1.66-1.58 (m, 4H), 1.57-1.43 (m, 4H), 1.09 (s, 3H)。
1.2.5 乙基(E)-3-(4-((1R,2R)-2-羟基-2-甲基环戊基)氨基)-2-((1-(甲基磺酰基)哌啶-4-基)氨基)嘧啶-5-基)丙烯酸酯(K-5)的制备
将K-4(24 g,53.7 mmol)溶于DMF(250 mL)中,依次加入碳酸钾(14.8 g,76 mmol),四三苯基磷钯(12.4 g,7.6 mmol)和丙烯酸乙酯(8 g,80.5 mmol)。氮气保护,升温至95 ℃,搅拌反应过夜。反应液冷却至室温后用硅藻土过滤,乙酸乙酯(200 mL)淋洗滤饼,滤液用水洗两遍,合并分液所得有机相,加入无水硫酸钠干燥、抽滤和减压浓缩后得到粗产物K-5(17.5 g),收率为70%。粗产物无需纯化可直接用于下一步。MS-ESI, m/z:468[M+H]+。1H NMR (400 MHz, DMSO-d6)δ:8.28 (s, 1H), 7.80 (d,J=15.6 Hz, 1H), 7.16 (d,J=28.3 Hz, 1H), 6.89 (s, 1H), 6.24 (d,J=15.6 Hz, 1H), 4.69 (d,J=51.8 Hz, 1H), 4.35 (s, 1H), 4.14 (q,J=7.1 Hz, 2H), 3.85 (s, 1H), 3.53 (d,J=6.7 Hz, 2H), 2.87 (s, 3H), 2.85-2.79 (m, 2H), 1.96 (d,J=23.9 Hz, 4H), 1.65 (s, 6H), 1.23 (t,J=7.1 Hz, 3H), 1.06 (s, 3H)。
1.2.6 8-((1R,2R)-2-羟基-2-甲基环戊基)-2-((1-(甲基磺酰基)哌啶-4-基)氨基)吡啶[2,3-d]嘧啶-7(8H)-酮(K-6)的制备
将K-5(17 g,36.4 mmol)溶于无水四氢呋喃(100 mL)中,于室温下加入1 mol/L的叔丁醇钾的四氢呋喃溶液(145.6 mL, 145.6 mmol),升温至45 ℃,搅拌至反应完成。反应液冷却至室温加水淬灭,反应液用乙酸乙酯稀释,分液得有机相,干燥、减压浓缩后得到粗产物,粗产物在正庚烷/乙酸乙酯的混合溶液(体积比10∶1)中打浆得到产物K-6(11.6 g),收率为76%。MS-ESI, m/z:422[M+H]+。1H NMR (400 MHz, CDCl3)δ:9.53 (s, 1H), 8.43 (s, 1H), 7.46 (d,J=9.3 Hz, 1H), 6.36 (d,J=9.3 Hz, 1H), 5.74 (s, 1H), 4.24 (s, 1H), 4.02 (s, 1H), 3.82 (s, 2H), 2.96-2.91 (m, 2H), 2.84 (s, 3H), 2.82 (s, 1H), 2.25 (d,J=3.0 Hz, 2H), 2.03 (dd,J=7.0, 3.2 Hz, 2H), 1.88-1.83 (m, 2H), 1.70-1.65 (m, 4H), 1.19 (s, 3H)。
1.2.7 8-((1R,2R)-2-羟基-2-甲基环戊基)-6-碘-2-((1-(甲磺酰基)哌啶-4-基)氨基)吡啶[2,3-d]嘧啶-7(8H)-酮(K-7)的制备
将K-6(11 g,26.1 mmol) 和N-碘代琥珀酰亚胺(8.8 g,39.1 mmol)溶于乙腈(110 mL)中,氮气保护,30 ℃搅拌30 min后,加入对甲苯磺酸水合物(0.5 g,2.6 mmol),继续氮气保护,30 ℃搅拌2 h。TLC监测反应完成,反应液用5%的亚硫酸钠水溶液(150 mL)淬灭,减压浓缩至剩余溶液约150 mL,然后于0 ℃下搅拌1 h;混合物抽滤,滤饼用5%的乙腈水溶液(150 mL)洗3次,置于真空干燥箱干燥过夜,得淡黄色固体11.1 g,收率78%。MS-ESI, m/z:548[M+H]+。1H NMR (400 MHz, DMSO-d6)δ:8.60 (d,J=14.7 Hz, 1H), 8.44 (s, 1H), 8.03 (d,J=7.3 Hz, 1H), 5.90 (s, 1H), 4.37 (s, 1H), 3.57 (d,J=13.9 Hz, 2H), 2.87 (s, 3H), 2.86-2.78 (m, 2H), 2.17 (s, 2H), 1.90 (d,J=38.7 Hz, 4H), 1.65 (s, 3H), 1.46 (d,J=11.4 Hz, 1H), 0.93 (s, 3H)。
1.2.8 PF-06873600的制备
反应瓶A:将叔丁醇钾(6.8 g,60.3 mmol)和CuCl(3 g,30.1 mmol)溶于DMF(36 mL)中,30 ℃搅拌15 min;随后加入(二氟甲基)三甲基硅烷(7.5 g,60.3 mmol),30 ℃搅拌30 min;加入K-7(11 g,20.1 mmol)和丙二醇(2.3 g,16.1 mmol)的DMF(30 mL)溶液,于30 ℃搅拌16 h。
反应瓶B: 将叔丁醇钾(6.8 g,60.3 mmol)和CuCl(3 g,30.1 mmol)溶于DMF(28 mL)中,30 ℃搅拌15 min;随后加入(二氟甲基)三甲基硅烷(7.5 g,60.3 mmol),30 ℃搅拌30 min。
将反应瓶B的混合溶液加入反应瓶A中,30 ℃ 搅拌20~72 h,TLC监测反应完成。反应液用2-甲基四氢呋喃(100 mL)转移至装有饱和氯化铵水溶液(60 mL)和35%氯化镁水溶液(60 mL)的反应瓶中,室温下搅拌30 min后分液,水相用 2-甲基四氢呋喃(60 mL)萃取一遍,合并有机相。有机相中加入丙酮(60 mL)后依次用饱和氯化铵水溶液(60 mL)和水(60 mL)洗两遍;混合溶液经硅藻土过滤得滤液,滤液减压浓缩和重结晶后得到白色固体,即PF-06873600(8.4 g),收率为89%。MS-ESI, m/z:470.2[M-H]-。1H NMR (400 MHz, DMSO-d6)δ:8.80-8.75 (m, 1H), 8.21 (s, 1H), 8.09-7.97 (brs, 1H), 7.01-6.74 (m, 1H), 5.85(brs, 1H), 4.41-3.90 (m, 2H), 3.58-3.41 (m, 6H), 2.84 (s, 3H), 2.35-2.33 (m, 2H), 2.19-1.49 (m, 7H), 1.04 (s, 3H)。
2 结果与讨论
2.1 起始原料的替换
以廉价易得的1-甲基环戊烯为起始原料,经一步反应即可得到较为昂贵的原研路线起始原料1-甲基-6-氧杂双环[3,1,0]己烷,且该步反应收率高达95%,这一变更极大地降低了合成成本。
2.2 重要中间体原料的替换
在合成吡啶并嘧啶母核结构时,原研路线使用了较为昂贵的原料(4-氯-2-(甲硫基)嘧啶-5-基)甲醇,本工艺路线中改用廉价的5-溴-2,4-二氯嘧啶,经三步反应即可得到关键中间体9(K-6),关键中间体9(K-6)的合成路线及收率见表1,由表1数据可以看出,本工艺路线较原研路线缩短了一步且收率提升至36.2%。
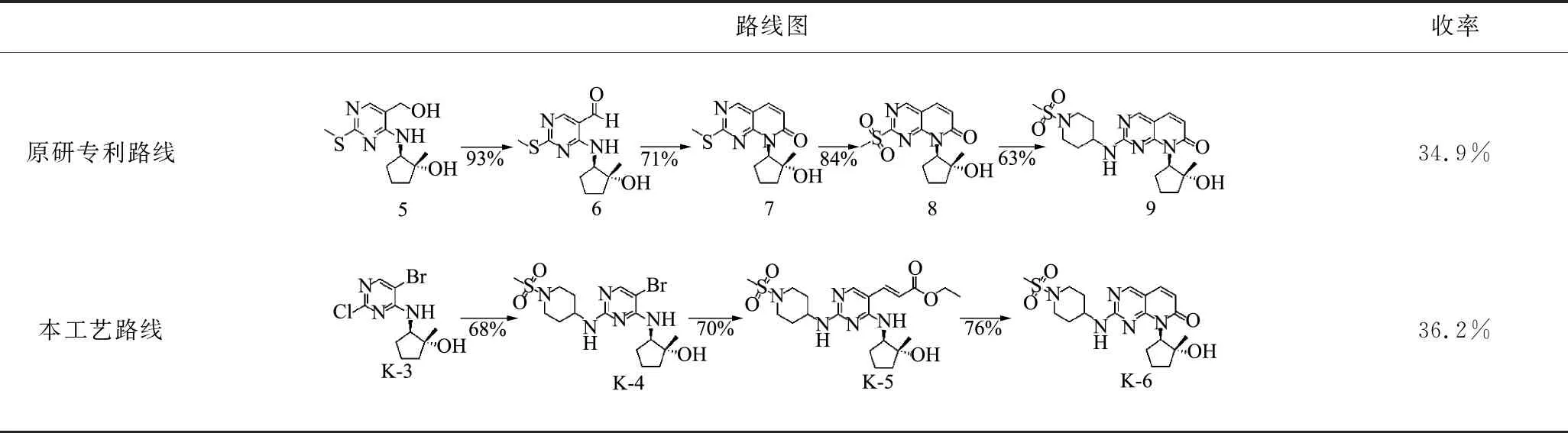
表1 关键中间体9(K-6)的合成路线及收率
2.3 中间体K-6的后处理优化
对中间体K-6进行后处理,发现K-6在正庚烷/乙酸乙酯的混合溶液体系中溶解性不佳,通过实验寻找最佳的二元溶剂配比(表2)。由表2可以看出,当正庚烷∶乙酸乙酯=10∶1时,K-6的纯度及收率达到最大值,因此在后处理时,选择正庚烷∶乙酸乙酯=10∶1作为打浆溶剂。

表2 不同比例的二元溶剂打浆对K-6纯度与收率的影响
3 结论
本文改进了PF-06873600的合成路线,用1-甲基环戊烯代替昂贵的1-甲基-6-氧杂双环[3,1,0]己烷作为起始原料,经还原、手性氨基酸的拆分、脱保护基和取代得到K-3,同时用5-溴-2,4-二氯嘧啶替代了4-氯-2-(甲硫基)嘧啶-5-基)甲醇,进一步降低了合成成本。中间体K-3经取代、Heck反应完成吡啶并嘧啶母核结构的合成,缩短反应步骤的同时也提高了收率。最后,关键中间体K-6经碘化和二氟甲基铜配合物得到目标化合物PF-06873600,纯度为98.5%。改进后的工艺路线总收率为12.4%。该方法使用的原材料简单易得、后处理操作简便,产物纯化多采用打浆重结晶,避免柱层析分离,便于放大生产,同时也为PF-06873600及其衍生物的合成提供参考。
