氧化亚钴表面CO活化解离反应机理的DFT计算
2022-08-31刘小浩
辛 蕾 刘 冰 姜 枫 刘小浩
(江南大学 化学与材料工程学院, 江苏 无锡 214122)
费托反应[1]是在催化剂及适当反应条件下将合成气(CO和H2)转化为长链碳氢化合物的工艺过程。自1920年以来,从最初使用煤作为原料生产合成气到使用生物质等替代,费托反应一直受到广泛关注。随着石油资源的日益短缺,以及合成气来源的多样化,由费托反应制备液体燃料和化学品受到人们越来越多的重视。在我国“贫油,少气,富煤”的能源结构和科学家们战略提出的“煤转油”的要求下,费托反应成为我国发展循环经济、实现石油替代的有效途径。目前用于费托反应的催化剂是多样的,但普遍认为金属是费托反应的活性中心,金属催化剂[2]如Fe、Co、Rh、Ru和Ni仍然是人们研究的重点和突破点,最受人们欢迎的是Fe基和Co基催化剂。其中Co催化剂[3]不容易产生积碳,产物分布以长链烷烃为主,相比于Fe催化剂,产物中CO2的选择性较低,较好地符合绿色化工、低碳环保的要求。
由于费托反应使用的催化剂往往是复杂的多组分化合物,活性相和非活性相直接清晰的界限判定起来有一定的困难。金属钴[4]被普遍认为是费托反应中的活性相;部分研究说明碳化钴[5]是一种表面物质,在CO的反应性和产物的选择性上有着较为显著的影响;此外,CoO[6]作为反应过程中的中间物种也受到了关注。1999年,Ernst等人[7]发现,当氧化态的钴存在时,对于C22+的选择性降低,C5-C13的选择性升高,认为氧化钴在费托反应中表现得非常活跃。据报道[8],当CoO负载在一些载体材料上,对费托反应和CO2加氢反应有较好的性能。Somorjai等[9]发现当CoO负载在TiO2上时,相比于金属Co,费托反应和CO2加氢表现得更加活跃。最近,有实验[10]评价了分散在SiO2载体上的混合CoO-Co纳米粒子对费托反应的作用。该催化剂中的CoO相能够促进费托反应和加氢反应。到目前为止,对于CoO的反应机理以及相关的理论研究非常少,因此,对此进行研究显得极其必要。
1 计算方法
本文通过使用VASP[11,12]软件包进行所有的密度泛函理论(DFT)计算。采用Perdew-Burke-Ernzerhof(PBE)泛函[13],PAW[14]方法用来近似描述原子核与外层电子之间的相互作用。截断动能设为400 eV。对于催化剂模型,构造了尺寸为a=b=9.03Å,c=21.39Å的(3×3)周期性CoO的slab模型,该模型包含72个原子。在表面顶部引入了15Å的真空层,以分离slab模型的底部和顶部。在优化过程中,CoO(200)、CoO(220)、CoO(111)的最底层原子层均被固定,K点分别设置为2×2×1、2×3×1、2×3×1。另外,对于金属Co催化剂模型,构造了尺寸为a=9.47Å,b=9.92Å,c=21.00Å的(2×4)周期性slab模型,该模型包含64个原子,在优化过程中,Co(10-11)的最底层原子层被固定,K点设置为2×2×1。10-5eV和0.02 eV/Å分别被用作电子自洽迭代和几何优化的收敛标准。采用CI-NEB[8,9]方法计算和搜索过渡态结构,通过计算是否是一个虚频检验过渡态是否准确。
2 结果与讨论
2.1 CoO(111)、(200)、(220)与Co(10-11)表面吸附物种结构
通过一系列吸附位点测试,选取各个晶面吸附能最强的结构进行后续计算,CO、HCO、H在CoO不同晶面和Co(10-11)面的吸附结构图如图1所示。
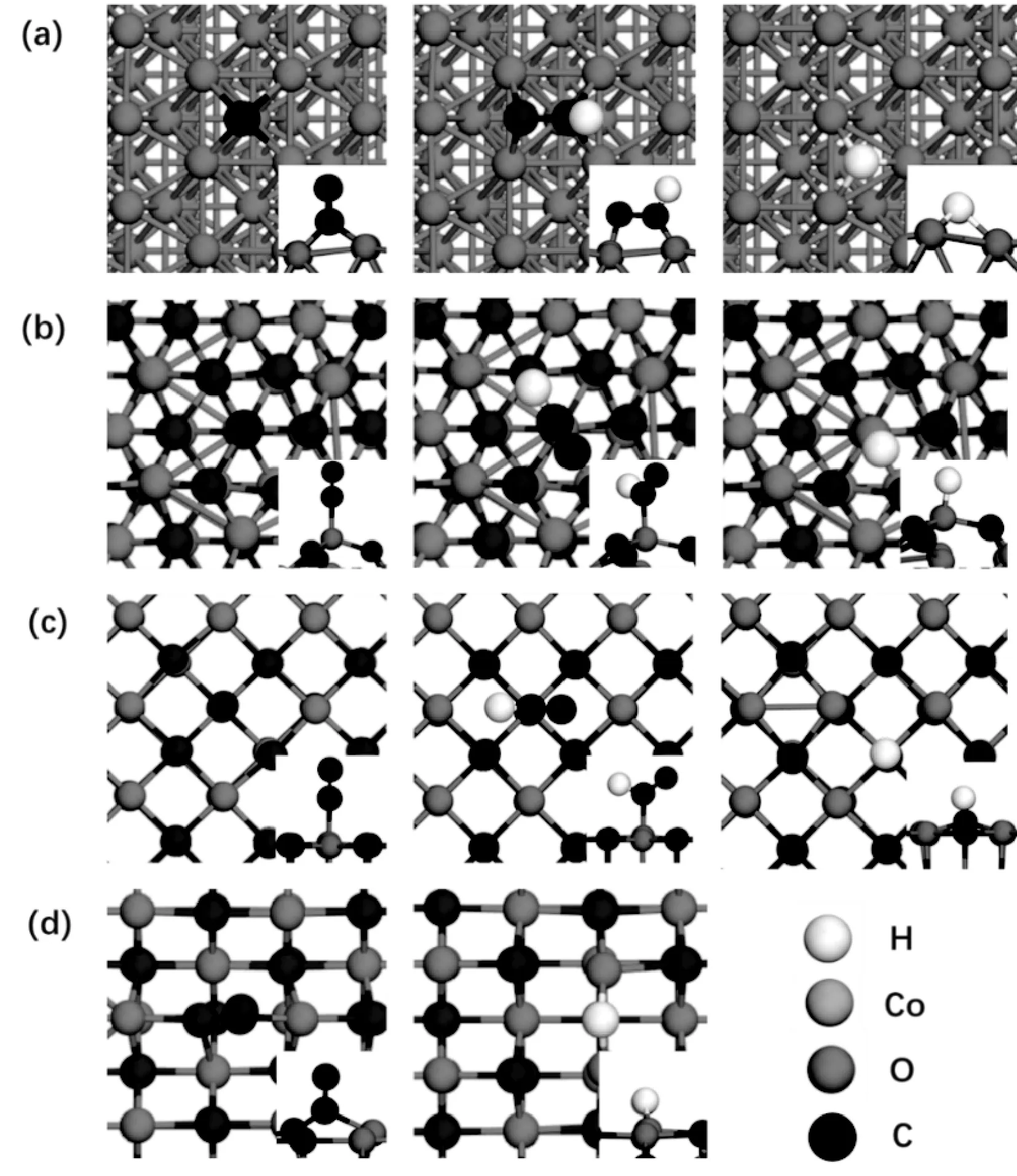
图1 (a) Co(10-11)面吸附CO、HCO、H的结构模型;(b) CoO(111)面吸附CO、HCO、H的结构模型;(c) CoO(200)面吸附CO、HCO、H的结构模型;(d) CoO(220)面吸附CO、HCO、H的结构模型
对于CoO(111)和(200)表面,一氧化碳与钴位点以单配位的形式吸附结合,HCO的吸附构型与一氧化碳类似,也是以单配位的形式与钴位点吸附结合,H也是以单配位的形式与钴位点吸附结合。而Co(10-11)表面上CO、HCO和H都是以多配位的吸附构型与钴位点吸附结合,这表明金属钴对CO和H具有更强的成键能力。
2.2 CoO(200)晶面CO活化解离机理
CO在CoO表面吸附后,C-O键的断裂一般考虑两种解离机理:H辅助CO解离和CO直接解离。由于CO直接解离非常困难,这里考虑H辅助解离CO路线。H辅助解离路径会有两种中间产物*HCO和*COH,这两种中间物种通过不同的解离路径进行。DFT计算能垒结果如图2所示,CO加H生成*COH 克服1.41 eV活化能垒,此过程吸热0.43 eV,而CO加H生成*HCO只需要克服0.09 eV的能垒,并且该过程放热1.07 eV,以上结果表明:相较于*COH 路径,CO和H反应生成*HCO物种在热力学和动力学上都更容易一些。生成的*HCO中间产物克服一个很低的能垒(0.24 eV)后,进一步通过H辅助解离路线生成*CH2OH物种,该基元反应克服0.55 eV 能垒,放热1.34 eV。最后*CH2OH物种克服1.72 eV能垒进行C-O键断裂,生成*CH2、*H和*OH这些费托反应的中间产物,进行后续催化剂上的加氢和偶联反应。从上可知,在CoO催化剂上,CO通过*HCO的路径解离比通过*COH路径解离更容易,*CH2OH物种解离生成*CH2、*OH和*H是该反应的速率控制步骤。
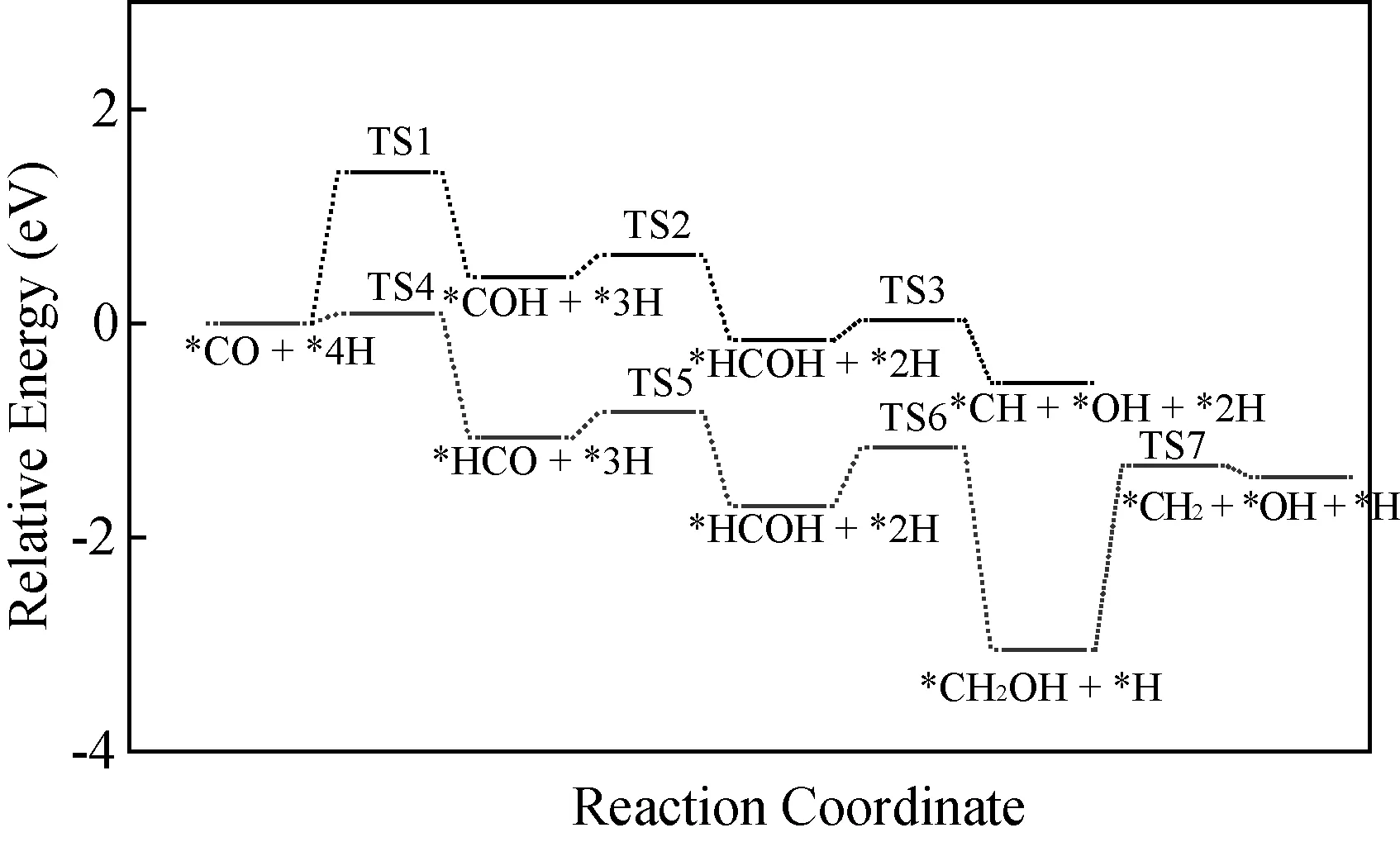
图2 CoO(200)表面上CO活化的反应能垒图
2.3 CoO(200)晶面CO活化解离的微观动力学研究
基于以上基元反应步骤和能量信息,构建微观动力学模型,通过求解微观动力学方程获得表面物种覆盖度的信息,如图3所示。在400 K时,表面主要为HCOH*和CH2OH*物种,随着温度升高,表面物种主要为CH2OH*,而CH2*、CH*等C-O键解离产物的覆盖度几乎为0,这表明CoO表面不利于C-O键的解离,这主要是由于CH2OH*物种在CoO表面上解离具有很高的能垒。

图3 CoO(200)表面CO活化解离的表面物种覆盖度
3 结论
密度泛函理论计算表明,金属Co(10-11)有着对CO、H和表面CHx物种更强的吸附,相比于Co的(10-11)面,CO在CoO的不同晶面上与钴位点以单配位的形式吸附。反应机理的计算结果表明当CO吸附在CoO(200)表面后可以通过H辅助路径生成*HCO物种,通过*HCO物种继续加氢生成*HCOH 和*CH2OH,然后解离生成*CH2、*H和*OH这些中间产物,*CH2OH物种的解离是该反应的速率控制步骤。微观动力学结果揭示了CO解离过程表面物种的覆盖度,表面物种主要为CH2OH*,这是由于CH2OH*物种在CoO表面上的解离具有很高的能垒。
