PMP22基因相关性周围神经病的遗传学和临床特点分析
2022-08-30朱啸巍詹飞霞刘时华栾兴华
朱啸巍,詹飞霞,张 超,刘时华,钟 平,曹 立,栾兴华
1.上海交通大学医学院附属第六人民医院神经内科,上海 200030;2.安徽医科大学附属宿州医院神经内科,宿州 234000
周围髓鞘蛋白22(peripheral myelin protein-22,PMP22)基因位于染色体17p11.2,长度为40 kb,由人类保守的6 个外显子组成。该基因的热点突变集中在CMT1A-REP 序列,由染色体17p11.2 近端和远端的2 个大小为24 kb 的序列组成,位于1.5 Mb 区域的两侧,该区多以重复突变或缺失突变为主[1]。
PMP22基因相关性周围神经病是一组以PMP22基因突变导致的遗传性周围神经病。PMP22基因突变主要有3种致病突变类型,包括PMP22重复突变、缺失突变及点突变。不同突变类型可导致不同疾病表型。PMP22重复突变可导致腓骨肌萎缩症1A 型(Charcot-Marie-Tooth type-1A, CMT1A; OMIM #118220);PMP22缺失突变可导致遗传性压迫易感性神经病(hereditary neuropathy with liability to pressure palsies,HNPP;OMIM#162500);PMP22基因点突变可导致CMT1A 或CMT1E(OMIM #118300)以及Dejerine-Sottas 综 合 征 (Dejerine-Sottas syndrome, DSS;OMIM #145900) 和Roussy-Levy 综 合 征(Roussy-Levy syndrome,RLS;OMIM #180800)等其他周围神经病[2-3]。在所有PMP22基因相关性周围神经病中,CMT1A 最常见(约占79%),其次为HNPP(约占16%),DSS约占3%,CMT1E约占2%[2]。
腓骨肌萎缩症(Charcot-Marie-Tooth,CMT)是一组临床表型类似的遗传异质性疾病,50%以上的CMT病例是CMT1A[4-5]。CMT1A 是一种常染色体显性疾病,通常是由染色体17p11.2 上1.5 Mb 区域不均等交叉的非等位基因同源重组所致。其主要临床特点是对称性、缓慢进行性肢体远端肌肉无力,肌肉萎缩,感觉减退,腱反射减弱或消失[5]。HNPP 呈常染色体显性遗传,其典型的临床表现是反复发作的急性无痛性、短暂性、压力性神经麻痹,与压迫或轻微牵拉有关[6]。也有PMP22基因突变相关的罕见表型,如DSS 与RLS,他们是CMT1A 的2 种变异表型[7-12]。经典的表型很容易识别,但非典型表型的诊断仍是一个巨大的挑战。
本研究通过分析59 例PMP22基因相关性周围神经病患者的临床和遗传学特点,旨在扩展PMP22基因相关性周围神经病临床表型及基因型,以期为疾病诊断及遗传咨询提供依据。
1 对象和方法
1.1 研究对象
从2006 年—2022 年上海交通大学医学院附属第六人民医院神经内科收治的162 例周围神经病患者中,经基因检测筛选出26 例PMP22基因相关性周围神经病先证者及其33 例家系成员患者作为研究对象。收集先证者及家系成员的临床资料,包括人口学数据、临床表现、电生理特点、病理学特点等。所有研究对象均签署知情同意书。本研究经上海交通大学医学院附属第六人民医院伦理委员会批准(伦理审批编号2021-219)。
1.2 基因检测
采集先证者及相关家系成员外周静脉血样本(3 mL),标准的苯酚-氯仿方案提取基因组DNA,应用多重连接依赖的探针扩增(multiplex ligationdependent probe amplification,MLPA) 检测PMP22基因大片段缺失、重复突变。对MLPA结果阴性的患者行全外显子测序(whole exon sequencing,WES),对发现PMP22基因突变的先证者行Sanger 测序及家系共分离验证。采用Phyre2 网站预测PMP22 的结构。采用University of California Santa Cruz(UCSC)数据库(https://genome.ucsc.edu)对测得的点突变位点予以不同物种保守性分析。根据突变类型不同,将PMP22基因相关性周围神经病患者分为3 组,即PMP22重复突变、PMP22缺失突变和PMP22点突变。
1.3 神经电生理检查
12 例PMP22重复突变和4 例PMP22缺失突变共16 例PMP22基因相关性周围神经病患者用肌电图诱发电位仪(KeyPoint Focus,丹麦丹迪)检测;检测内容包括双侧正中神经、尺神经、胫神经和腓总神经或腓肠神经的运动神经传导速度(motor nerve conduction velocities,MCV)、复合肌肉动作电位波幅(compound muscle action potentials,CMAP)、感觉神经传导速度(sensory nerve conduction velocities,SCV);检测结果由至少2 位神经电生理专业医师评估。正中神经刺激点选择腕-拇短展肌(abductor pollicis brevis,APB)与肘-腕,尺神经刺激点选择腕-小指展肌(abductor digiti minimi,ADM)与肘下-腕,胫神经刺激点选择踝-拇展肌(abductor hallucis,AH),腓总神经或腓肠神经刺激点选择踝-趾短伸肌(extensor digitorum brevis,EDB)。比较PMP22重复和缺失突变患者正中神经、尺神经、胫神经和腓总神经或腓肠神经双侧的MCV、CMAP、SCV 差异是否具有统计学意义,并分析PMP22重复和缺失突变2组间上述指标的差异。
1.4 病理学检查
12 例PMP22基因相关性周围神经病患者行腓肠神经活检,其中2 例行腓肠神经和肌肉联合活检。周围神经组织石蜡包埋,肌肉组织液氮及异戊烷冷冻,7 μm 切片。切片经苏木精-伊红(hematoxylin-eosin,H-E)染色、改良Gomori 三色法(modified gomori trichrome,MGT)染色、刚果红染色,以及CD3 抗体(za-0503,北京中杉金桥生物技术有限公司;1∶200)、CD20 抗体(ab-64088,美国Abcam 公司;1∶1 000)和CD68抗体(zm-0464,北京中杉金桥生物技术有限公司;1∶200)免疫组织化学(免疫组化)染色。神经透射电镜标本在2.5%戊二醛和1%锇酸中固定,然后嵌入环氧树脂。用柠檬酸铅电子染色后,电子显微镜观察超薄切片。
1.5 统计学分析
采用SPSS 26.0 软件进行数据分析。定量资料先通过K-S检验判断正态性,正态资料用±s表示并采用独立样本t检验,偏态资料用M(P25,P75)表示并采用Mann-Whitney 检验。P<0.05 表示差异具有统计学意义。
2 结果
2.1 遗传学特点
通过MLPA、全外显子测序和Sanger 测序发现PMP22基因突变的周围神经病患者共59 例,包括13个家系患者共46 例,和13 例散发病例。其中PMP22重复突变51 例(86.4%),诊断为CMT1A;其主要来自12个家系(图1A),余7例为散发病例。PMP22缺失突变6 例(10.2%),诊断为HNPP;其中2 例来自1个家系(图1B),余4 例为散发病例。PMP22杂合点突变2例(3.4%),突变位点分别为c.C215T(p.S72L)和c.C215T(p.G100V)(图1C~E),诊断为DSS。根据2015 年美国医学遗传学与基因组学学会基因变异解读标准和指南(American College of Medical Genetics and Genomics, ACMG)[13], p. S72L 和p.G100V 均为“可能致病”(likely pathogenic),2 例PMP22点突变位点在不同物种高度保守(图1F)。

图1 家系图及基因突变分析Fig 1 Family pedigrees and analysis of gene mutation
2.2 临床特点
2.2.1 人口学数据 59 例PMP22基因异常的患者中,男性37 例,女性22 例,男女比例1.7∶1。就诊时年龄40.0(23.0,47.0)岁,发病年龄34.0(14.0,49.5)岁,发病年龄小于10 岁者7 例,从发病到确诊病程6.000(2.750,14.750)年。
在26 例PMP22基因相关性周围神经病先证者中,19 例PMP22重复突变先证者的发病年龄为40.0(25.0,49.0)岁,发病年龄小于10岁者3例,从发病到此次确诊病程5.00(3.50,8.50)年。5 例PMP22缺失突变先证者的发病年龄为15.0(14.0,24.0)岁,从发病到此次确诊病程0.088(0.034,2.103)年。2例PMP22点突变先证者均于出生后数月发病。从发病到此次确诊病程分别为12与20年。
2.2.2 临床表现 对先证者的临床症状进一步分析(图2)。在19 例PMP22重复突变先证者中,首发症状为肢体无力者13 例(68.4%)、肢体麻木3 例(15.8%)、肢体无力合并肢体麻木3 例(15.8%)。PMP22重复突变先证者中存在四肢末梢型感觉障碍7例(36.8%)、双上肢肌力减退11 例(57.9%)、双下肢肌力减退16 例(84.2%),以肢体远端握力减退或足背屈无力为常见表现。11 例(57.9%)患者存在不同程度的肌肉萎缩,以双手骨间肌萎缩多见,严重者双膝关节以下肌肉极度萎缩,呈“鹤腿样”。16 例存在四肢腱反射减低(84.2%)、5 例存在双手震颤(26.3%)、1 例出现双眼轻度短暂水平眼震(5.3%)。跨阈步态5 例(26.3%)、弓形足畸形6 例(31.6%),足内翻畸形2 例(10.5%)。所有PMP22重复突变患者未出现爪形手畸形、脊柱侧弯畸形、共济失调等临床症状。
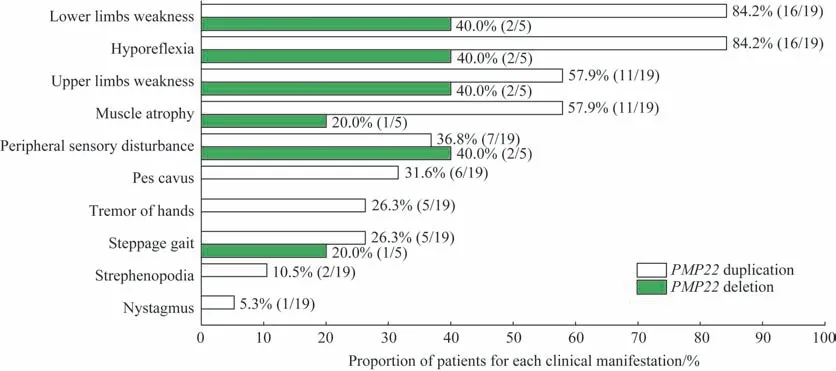
图2 PMP22基因重复突变和缺失突变先证者的临床特点Fig 2 Clinical features of probands with PMP22 duplication and deletion mutations
在5 例PMP22缺失突变的先证者中,4 例(80.0%)患者因足下垂就诊、1例(20.0%)患者因右手麻木无力就诊,患者症状较轻。存在四肢末梢型感觉障碍2例(40.0%)、双上肢肌力减退2例(40.0%)、双下肢肌力减退2 例(40.0%),出现右手虎口、大小鱼际、骨间肌肌肉萎缩1 例(20.0%)。四肢腱反射减低2例(40.0%)、跨阈步态1例(20.0%)。
2 例PMP22点突变患者的首发症状为四肢无力,病程长,临床症状重,均存在肌肉萎缩、四肢肌力减退、腱反射消失。其中1 例患者有马蹄足畸形和四肢末梢型感觉减退,另1 例患者有严重的脊柱侧弯畸形。
2.2.3 电生理特点 16 例PMP22基因相关性周围神经病患者进行神经电生理检测,均以脱髓鞘病变为主。与正常参考值相比,12 例PMP22重复突变的患者MCV 明显减慢,SCV 减慢或未引出,CMAP 波幅明显降低或未引出。与正常参考值相比,神经传导速度减低及CMAP 波幅下降多呈对称性,部分患者肌电图可见纤颤波。PMP22缺失突变的4位先证者肌电图呈脱髓鞘性周围神经病变,可伴有轴索损害(50.0%),以单一运动神经局部脱髓鞘为主要改变。其中,尺神经MCV 减慢者占50.0%,CMAP 波幅下降者占37.5%;腓总神经MCV 减慢者占87.5%,CMAP 波幅下降者占25.0%,提示神经传导阻滞。正中神经SCV 均减慢,波幅下降者占50.0%;尺神经SCV均减慢,波幅均下降。
与PMP22缺失突变患者相比(表1),PMP22重复突变患者在左右两侧的正中神经、尺神经、胫神经和腓总神经的MCV、CMAP、SCV 明显降低,差异具有统计学意义(P<0.05)。在PMP22的2 种突变表型中,左右肢体MCV、CMAP、SCV 未见明显差异(P>0.05),表现为对称性神经病变。
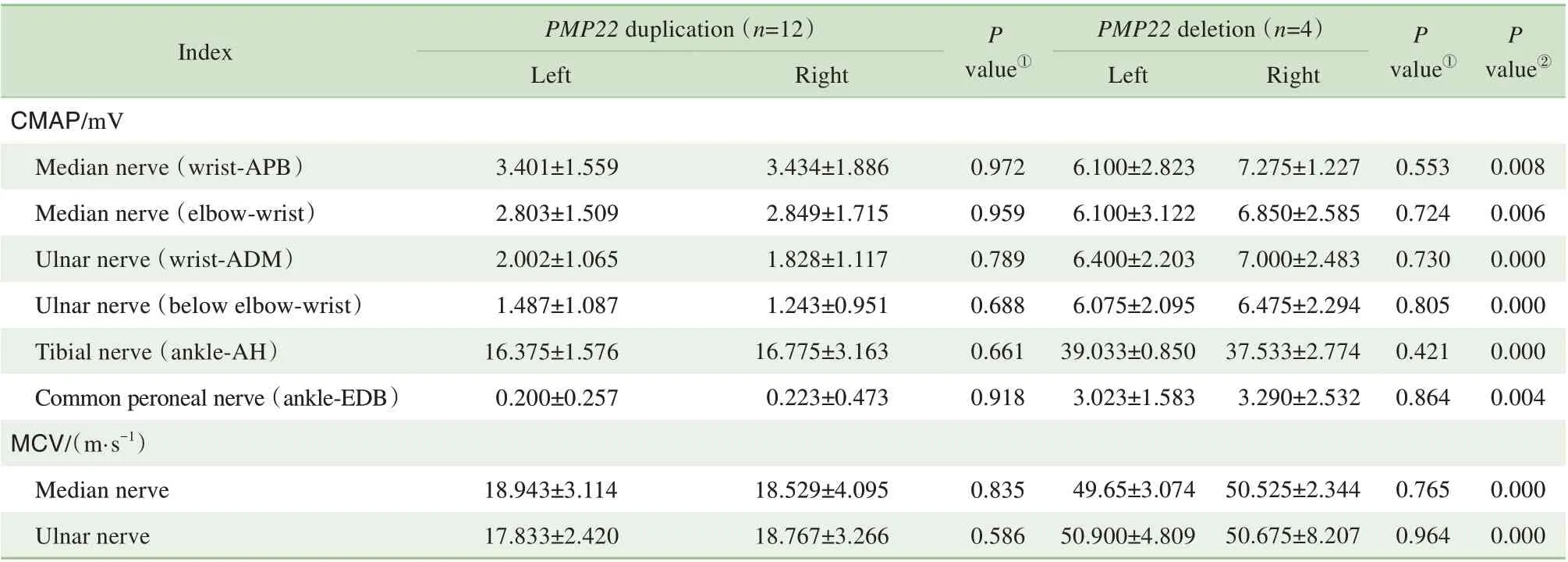
表1 PMP22重复突变和缺失突变患者的电生理参数比较Tab 1 Comparison of electrophysiological parameters between PMP22 duplication group and PMP22 deletion group
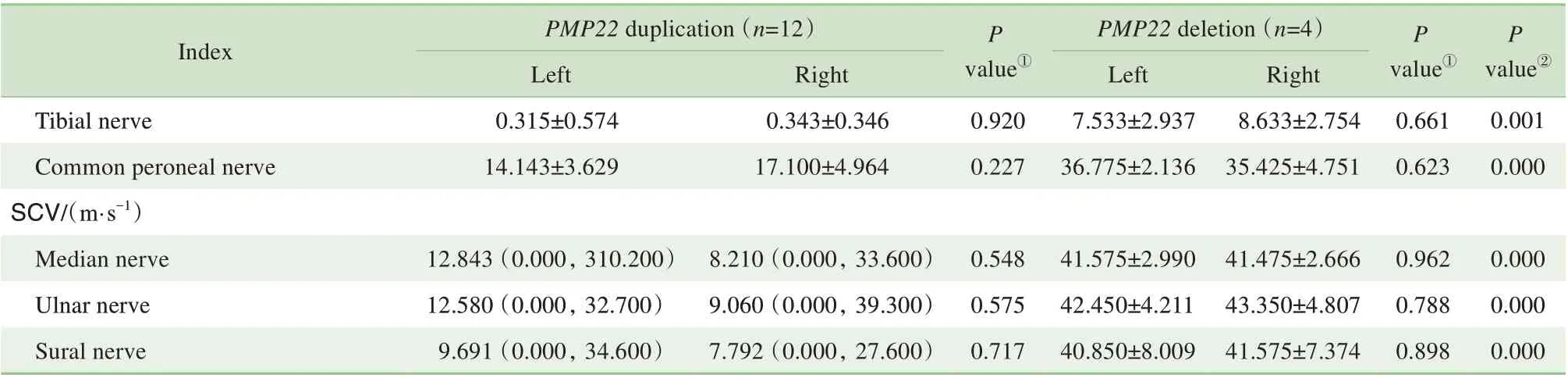
Continued Tab
2.2.4 病理特点 对8 例PMP22重复突变、3 例PMP22缺失突变和1 例PMP22点突变患者行腓肠神经活检,其中2 例PMP22重复突变患者行腓肠神经和腓肠肌联合活检。在光学显微镜下,MGT 染色可见有髓神经脱髓鞘,H-E 及CD3、CD20、CD68免疫组化染色未见明显炎性细胞浸润,也未见淀粉样物质沉积。透射电镜下观察,8 例PMP22重复突变患者腓肠神经活检可见髓神经纤维密度程度降低,伴大量有髓神经纤维的“洋葱球样”结构出现,部分患者的无髓神经纤维密度减少(图3A)。2 例PMP22重复突变的腓肠肌活检可见散在和成组分布的小角状萎缩肌纤维,呈群组化现象,提示神经源性肌肉病变。3 例PMP22缺失突变患者腓肠神经活检可见有髓神经纤维密度轻度降低,伴个别有髓神经纤维髓鞘增厚(图3B)。1 例PMP22点突变患者腓肠神经活检可见慢性重度脱髓鞘性周围神经病病理改变,也见大量“洋葱球样”有髓神经纤维。
3 讨论
PMP22基因相关性周围神经病是一组因PMP22基因突变所导致的遗传性周围神经病,其临床表现和严重程度在同一家族成员或不同家系间存在差异;一些家族成员临床无症状,仅在家系筛查时被诊断[2,14]。与PMP22重复突变的患者相比,PMP22缺失突变的HNPP 患者症状相对较轻,发病早,病程较短[6,15]。仅有少数患者有严重残疾,见于PMP22点突变患者。既往研究[16]表明,PMP22点突变比重复突变或缺失突变引起的临床症状更为严重,可表现为DSS 表型。疼痛是CMT1A 患者常见的主诉,55%~70%的CMT1A 患者会出现疼痛,主要局限于足部,呈神经性或肌肉性疼痛[17]。但在本研究中,仅有1例PMP22重复突变患者(散发病例)在病程中出现疼痛。疼痛的病因并不明确,可能与病程、行走的姿势异常、感觉或自主神经病变等因素有关。PMP22缺失突变所致的HNPP 表型以感觉运动性神经病最多见,通常无疼痛表现。有2 例PMP22缺失突变的HNPP 患者,临床表现不典型,在基因检测诊断之前拟诊断为CMT,说明HNPP 表型具有明显的临床异质性,易被误诊。
PMP22 蛋白在周围神经中高度表达,是髓鞘磷脂的跨膜糖蛋白成分,具有协调髓鞘磷脂的功能。PMP22 蛋白通过适当的结构折叠和调节参与维持Schwann 细胞的稳态[18-20]。PMP22基因剂量效应是导致CMT1A 和HNPP 的重要机制。CMT1A 患者皮肤活检、腓肠神经活检中的PMP22mRNA 和蛋白水平升高,而HNPP 患者上述水平降低,支持了这一观点[18-21]。染色体内和染色体间的作用也可以调节基因的结构和表达。PMP22重复的DNA 可能会打乱正常基因组的结构和表达,导致PMP22转录不稳定,引起PMP22 表达的失调,这可以解释一些PMP22基因重复突变患者即使PMP22mRNA 和蛋白水平处于正常范围内,仍然存在典型CMT1A 的临床症状[22]。在HNPP 患者中,PMP22 蛋白缺乏会导致连接蛋白复合体移位,破坏髓鞘间的紧密连接和黏着连接,致使鞘磷脂过度渗透,髓鞘通透性异常增加,抑制动作电位的传递,导致可逆性神经传导阻滞;可逆性神经传导阻滞是HNPP 临床症状反复缓解、复发的基础[23]。
本研究2 例PMP22点突变的患者均为DSS 表型,患者分别携带p.S72L 和p.G100V 突变。既往曾有p. S72P 和p. S72W 突 变 所 致DSS 的 报 道[24-25]。p.G100V 突变则可表现为DSS 和CMT 2 种临床类型[11-12]。DSS是CMT1疾病谱中的一种特殊表型,其发病早、症状重、进展快,主要的遗传方式为常染色体隐性遗传,偶有常染色体显性遗传。PMP22点突变导致的表型可从轻度的HNPP表型到DSS和先天性髓鞘形成障碍性神经病(congenital hypomyelinating neuropathy,CHN) 等重度早发性髓鞘障碍神经病[26-27],具有高度临床异质性。
神经电生理是评价神经功能的有效检查手段。本研究显示MCV、CMAP、SCV在左右肢体中差异未见统计学意义,表现为对称性神经病变[28]。PMP22重复突变患者的左右正中运动神经平均MCV分别为(18.943±3.114)和(18.529±4.095)m/s,与既往研究结果(约为20 m/s)接近[29]。个别CMT1A患者出现了神经传导阻滞,表现为对称性多部位神经传导阻滞;而HNPP患者表现为局灶性运动和SCV减慢,以尺神经多见。本研究发现2例临床无症状者,经电生理检查发现脱髓鞘病变,通过PMP22基因检测确诊,提示在先证者家族成员中可优先选择电生理评估神经传导速度。
PMP22基因重复突变可导致髓鞘厚度增加、反复脱髓鞘和髓鞘再生,逐渐形成经典的“洋葱球样”结构[30]。PMP22缺失突变患者的腓肠神经活检呈局灶性髓鞘增厚和节段性脱髓鞘,纵切面可见局部髓鞘增厚区域隆起,形似腊肠,故称作“腊肠样”结构[31]。PMP22点突变患者可出现类似PMP22重复突变形成的“洋葱球样”结构,从病理上不能将二者进行区分[32]。广泛的有髓神经“洋葱球样”改变是CMT1A 患者的病理特征,可在患儿6 岁后逐渐形成,可与其他周围神经病鉴别[33]。本研究部分PMP22重复突变患者(散发病例)的病理结果显示有继发性轴突变性表现。继发性轴突变性是脱髓鞘神经病变的常见特征,这一过程常与患者的行走异常和长期残疾有关[34]。在PMP22缺失突变患者腓肠神经中可见部分明显增粗的有髓神经纤维,符合髓鞘肥厚性周围神经病的病理改变。髓鞘缩短和异常增厚可导致HNPP 神经传导速度减慢,临床表现严重的青年患者肌电图也可出现神经传导速度显著减慢[35]。
总之,本研究总结了PMP22基因相关性周围神经病的临床特征,包括发病年龄、严重程度、家族内和家系间的异质性、神经电生理特点及病理特点。临床疾病谱的变化可能受到PMP22基因不同突变的影响,PMP22重复突变导致CMT1A,PMP22缺失突变导致HNPP,PMP22点突变导致DSS。对于神经电生理和病理检查呈脱髓鞘表现的患者,排除获得性病因后,应予以PMP22基因拷贝数变异和点突变的基因检测;对于家族中无症状者,可行神经传导速度筛查或PMP22基因检测,从而有助于该病的优化诊断以及遗传咨询。
利益冲突声明/Conflict of Interests
所有作者声明不存在利益冲突。
All authors disclose no relevant conflict of interests.
伦理批准和知情同意/Ethics Approval and Patient Consent
本研究已通过上海交通大学医学院附属第六人民医院伦理委员会的审核批准(文件号2021-219)。受试对象或其亲属已经签署知情同意书。
All experimental protocols in this study were reviewed and approved by the Ethics Committee of Shanghai Sixth People's Hospital,Shanghai Jiao Tong University School of Medicine (approval letter No. 2021-219). Consent letters have been signed by the research participants or their relatives.
作者贡献/Authors'Contributions
曹立、钟平、栾兴华参与了研究设计;朱啸巍、詹飞霞参与了论文的写作和修改。所有作者均阅读并同意了最终稿件的提交。
The study was designed by CAO Li, ZHONG Ping and LUAN Xinghua. The manuscript was drafted and revised by ZHU Xiaowei and ZHAN Feixia. All the authors have read the last version of paper and consented for submission.
·Received:2021-12-06
·Accepted:2022-05-06
·Published online:2022-05-28
