土家族脊髓小脑共济失调一家系报道
2022-08-26熊小平王荣耀李渝田小龙杨章琴钟敏
熊小平 王荣耀 李渝 田小龙 杨章琴 钟敏
1 病例报告患者(先证者),女,土家族,51岁。因“进行性步态不稳20余年,伴言语不清5年”入作者医院。患者于30余岁时开始出现双下肢步态不稳,有踩棉花感及踏空感,走路摇晃,不能走直线。病情进行性加重,渐出现双下肢独立行走困难,需拄杖行走。5年前出现言语笨拙,吐字不清,饮水呛咳,无吞咽困难,症状呈进行性加重。目前患者基本卧床,尚能勉强扶走,言语含混不清。病程中有多汗、记忆力下降。查体:血压125/80 mmHg,身高1.57 m,有轻微胸椎后突。简易智能精神状态检查量表(MMSE)19分,双眼球内收、外展及上视均略差,有水平眼震,双瞳孔等大等圆,光反射灵敏。双侧鼻唇沟对称,伸舌居中,舌肌无萎缩,无舌肌震颤。咽反射存在,言语含糊不清。颈软。行走困难,需扶走,双下肢肌肉略萎缩,四肢肌力和肌张力尚可。浅感觉正常,深感觉障碍,深反射活跃,浅反射正常,指鼻试验及跟-膝-胫试验欠稳准,双侧巴宾斯基征阳性。辅助检查:血常规、肝肾功能、血糖、血脂检测均正常。患者左右眼底摄片检查:双眼视网膜均未见明显异常,眼底血管未见明显异常。影像学检查见小脑、脑干及脊髓均见明显萎缩(图1)。

图1 先证者(A、B)及Ⅲ10(C、D)患者头部及颈髓MRI表现:头颅MRI矢状位T2加权像示小脑、脑干萎缩短(A),颈髓MRI矢状位T2加权像加权像示颈髓萎缩(B);头颅MRI矢状位T2加权像示小脑、脑干萎缩(C),颈髓MRI矢状位T2加权像加权像示颈髓萎缩(D)
先证者样本致病基因ATXN3(CAG)拷贝数为15/64,发生异常扩增(北京迈基诺医学检验所检测,CAG扩增拷贝数>44考虑为脊髓小脑共济失调)(图2)。该结果支持受检人为脊髓小脑共济失调3型(spinocerebellar ataxias 3,SCA3)。诊断:SCA3。

图2 先证者基因检测(PCR+片段分析检测方法)结果:SCA3基因(CAG)拷贝数为15和64,发生异常扩增(箭头所指处为异常增张拷贝)
本家系为土家族家系,家系中Ⅰ、Ⅱ代均与土家族通婚,Ⅲ代中除Ⅲ13、Ⅲ14与汉族通婚,余均与土家族通婚。Ⅳ代中,Ⅳ4、Ⅳ8与汉族通婚,Ⅳ5、Ⅳ9及Ⅳ15均与土家族通婚。
该家系共有5代49人,其中患者16例,男、女各8例。先证者(Ⅲ4)外婆(Ⅰ1)发病年龄不清,死亡年龄68岁;家系中第二代患者发病年龄均在40岁左右,约60岁死亡。先证者的弟弟(Ⅲ5)发病年龄为27岁,约46岁死亡;先证者表妹(Ⅲ8)发病年龄在28岁,43岁死亡;先证者的表弟、表妹( Ⅲ9、Ⅲ10、Ⅲ11、Ⅲ14)发病年龄均为30余岁,现年30~40岁,其中Ⅲ8与Ⅲ10为近亲结婚,其子为Ⅳ11,5岁发病,11岁死亡;Ⅲ8与Ⅲ10离婚后,Ⅲ10再婚育Ⅳ13、14。先证者表侄Ⅳ12,8岁发病,13岁死亡。第五代尚未发现患者。家系中患者的病情严重程度不一,但均主要表现为共济失调、深感觉障碍、舌肌震颤、眼球运动障碍、突眼及眼震,病程后期伴有认知功能障碍,且有明显的遗传早现现象(图3、表1)。
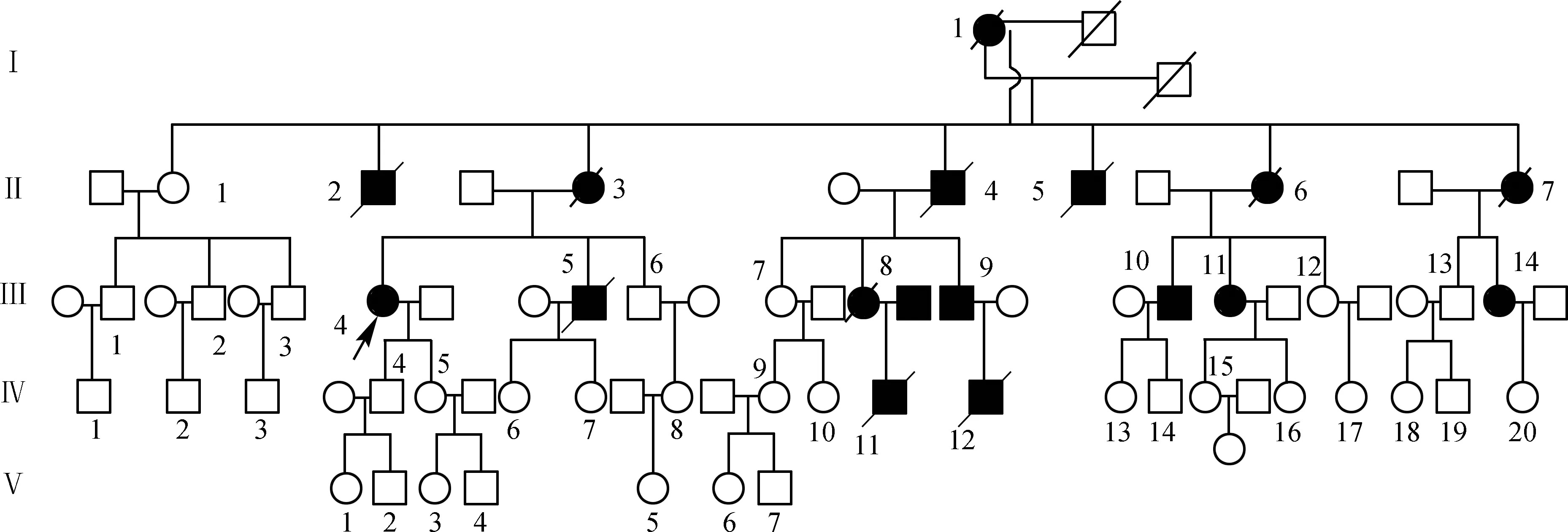
注:□:正常男性;■:男性患者;○:正常女性;●:女性患者;↗:先证者;/:死亡者 图3 患者家系图谱

表1 不同亲代发病情况
本次研究共收集家系成员44人的临床信息,根据疾病的遗传模式分析及成员的知情同意,共收集10位家系成员血液行基因检查。其中,第Ⅱ代家系成员1位(即Ⅱ1),无临床表现,基因检测ATXN3(CAG)拷贝数为9。第Ⅲ代家系成员共7位,4位为患者(Ⅲ4、Ⅲ9、Ⅲ10、Ⅲ11),有临床症状,其基因检测结果提示致病基因ATXN3(CAG)拷贝数为64~67;其余3位(Ⅲ1、Ⅲ3、Ⅲ7)基因检测ATXN3(CAG)拷贝数正常,分别为20、9、22。第Ⅳ代家系成员共2名(Ⅳ13、Ⅳ14),目前均无症状,为无症状致病基因携带者,基因检测结果提示致病基因ATXN3(CAG)拷贝数分别为70、68次(表2)。

表2 本家系患者及携带者临床表现、体格检查、基因检查一览表
2 讨论SCAs是一组神经遗传性疾病,为常染色体显性遗传,以平衡障碍、共济失调及言语含混为临床特征。目前,已发现有47个SCAs亚型,其中35个致病基因已明确。由于A致病基因的CAG重复序列持续扩增突变,其基因编码产物多聚谷氨酰胺增多,在神经组织内异常聚集,从而引发疾病[1]。目前报道的中国SCA患者中,SCA3亚型是最常见的一种,超过50%[2]。
SCA3临床表现较为复杂,主要有共济失调,眼球运动障碍、锥体束征、锥体外系征、肌萎缩、视神经萎缩、帕金森、周围神经病、言语不清和痴呆等表现。根据临床表现分为4型。本家系患者主要表现为共济失调,眼球运动障碍、突眼、锥体束征、锥体外系征、言语不清和痴呆。第二代及第三代发病时间基本在30~40岁,死亡时间在50~60岁,病程进展相对缓慢,但遗传早现现象十分明显,其第四代发病基本上为6~9岁发病,病程进展快,13岁左右死亡。其临床表现符合Ⅰ型及Ⅱ型混合型。
据报道,在不同种族、不同地区SCA3患者临床表现有不同特点,致病基因ATXN3(CAG)重复次数存在差异。国外最低重复次数为45次,而国内为43次。目前重庆报道汉族本病ATXN3(CAG)重复次数为56~71次[3]。但土家族人该病发病特征及基因特点未见报道。一项针对贵州土家族15个STR基因座多态性研究报道认为,贵州土家族与湖北土家族基因最近,与同省汉族较近,表明土家族与其他民族基因仍存在差异。本家系为土家族家系,家系中Ⅰ、Ⅱ代均与土家族通婚,Ⅲ代中仅Ⅲ13、Ⅲ14与汉族通婚,余均与土家族通婚。Ⅳ代中,Ⅳ4、Ⅳ8与汉族通婚,Ⅳ5、Ⅳ9及Ⅳ15均与土家族通婚。家系患者ATXN3(CAG)重复次数为64~70次,健存患者中均为高拷贝数,拷贝数均>64,均符合SCA3诊断标准〔ATXN3(CAG)拷贝数>44〕,是否与土家族民族特征有关,尚待更多家系及患者基因分析和研究以阐明。本家系不同亲代患者该致病基因重复拷贝数有逐渐增加趋势,且临床上也可见Ⅲ代发病年龄较Ⅱ代患者发病年龄更早,病情更严重,提示遗传早现,其原因为Ⅲ代该致病基因拷贝数更高。Ⅳ13目前10岁,Ⅳ14目前13岁,尚无症状,但其ATXN3(CAG)重复次数分别为70、66次,其无症状可能与其未到发病年龄有关,可随访观察。ATXN3(CAG)重复次数<44次家族成员及其后代均未患病。Ⅱ7因亲属部同意未能检测基因,对其进行了遗传风险咨询及生育指导,嘱其生育后代之前应进行本家系中发现的致病基因验证,以最大程度避免家系内类似病例的发生。
目前SCA尚缺乏有效治疗手段,使用加巴喷丁等药物治疗及康复理疗仅可部分缓解患者的症状及改善生活质量。近年来,对该病遗传学和病理机制的深入研究,发现了可减缓疾病进展的潜在治疗靶点。潜在靶点的治疗方法可以分为针对下游毒性产物效应的药物治疗、基因治疗和干细胞替代治疗,仍需进一步研究。
志谢:感谢重庆医科大学附属儿童医院神经疾病诊治中心钟敏博士对本家系调查研究的指导。
