E3泛素连接酶在阿尔茨海默病中的作用机制研究进展
2022-08-26张晶晶王亚琦王培昌
张晶晶 王亚琦 王培昌
阿尔茨海默病(Alzheimer disease,AD),又称老年性痴呆,是一种以进行性认知能力下降,记忆力衰退并伴有人格改变为主要临床表现的神经系统退行性病变。AD主要影响老年人,由于人口老龄化,目前全世界约有4400万人患有AD。预计到2050年,患该病人数将达目前两倍以上,这将给人类、社会和经济带来巨大的负担[1]。尽管经过了几十年的研究,目前仍无有效逆转AD进程的方法[2]。AD患者的病理特点包括脑内大量的神经元凋亡和神经炎症,以及细胞外沉积的老年斑(senile plaques,SPs)和细胞内的神经纤维缠结(neurofibrillary tangles,NFTs)。SPs的主要成分是β-淀粉样蛋白(amyloid-β,Aβ),来源于淀粉样前体蛋白(amyloid precursor protein,APP)。NFTs是由于Tau蛋白过度磷酸化所产生的。AD的病因十分复杂,关于其疾病机制的理论学说众多。虽然到目前为止AD 发病的确切机制尚未阐明,但近年来研究人员发现在 AD 患者脑中不溶性蛋白质的积聚可能与泛素-蛋白酶体系统(ubiquitin-proteasome system,UPS)密切相关。此外,AD初期患者脑内蛋白降解缺陷会引起突触功能障碍,UPS在突触的正常活动中也起着关键作用[3]。
UPS又称泛素-蛋白酶体途径(ubiquitin-proteasome pathway,UPP),是一种常见的翻译后调控机制,负责正常和病理状态下蛋白质的降解。研究表明,由蛋白酶体介导的清除神经系统中错误折叠或损伤的蛋白质对维持细胞内稳态具有重要意义[4],并且在调控细胞周期和细胞凋亡中也起着关键作用[5-6]。同时,泛素信号也参与许多细胞过程,如自噬、信号转导、蛋白质的定位等[7]。更重要的是,最近的研究表明,UPS功能障碍与AD密切相关[8]。UPS主要由泛素(ubiquitin)以及一系列酶组成,主要包括泛素激活酶(ubiquitin-activating enzymes,E1s)、 泛素结合酶(ubiquitin-conjugating enzymes,E2s)和E3泛素连接酶(E3 ubiquitin ligases)以及26S蛋白酶体[9]。泛素是真核细胞中高度保守的一种修饰分子,由76个氨基酸组成,通过一系列酶促反应(包括 E1s、 E2s和 E3泛素连接酶)共价结合并标记靶蛋白底物。随后,在大多数情况下,修饰的底物蛋白被26S蛋白酶体复合物识别并降解[10]。其中E3泛素连接酶既可以决定靶蛋白的特异性识别,又可以作为桥梁将活化的泛素分子从E2s直接转移到靶蛋白,是整个泛素化过程中最为关键的酶。因此,本文将对E3泛素连接酶在AD发病中的作用机制的最新研究进展进行综述。
1 泛素化与E3泛素连接酶
1.1 泛素化泛素化是一种重要的蛋白质翻译后修饰,其通过对蛋白质稳定性、定位、活性以及相互作用的调控,广泛参与了各种生理过程,包括细胞增殖、凋亡、内吞、DNA损伤修复以及免疫应答等[11]。泛素化是通过E1s、E2s和E3泛素连接酶的连续级联反应实现的。首先,在ATP供能的情况下E1s的半胱氨酸残基与泛素C端赖氨酸残基之间形成高能硫酯键,从而活化泛素分子。然后,激活的泛素与E2s的半胱氨酸残基连接。最终,激活的泛素在E3泛素连接酶的作用下被转移到靶蛋白上。泛素分子全长包含7个赖氨酸残基(Lys6、 Lys11、 Lys27、 Lys29、 Lys33、 Lys48和 Lys63)、1个N端甲硫氨酸残基(Met1)和1个C 端的甘氨酸残基(Gly),被泛素化的蛋白质因其连接泛素的数量不同以及多聚泛素链位点的不同,经不同信号途径处理,功能也不尽相同[9]。如果只有单一泛素分子被转移到靶蛋白上,则称为单泛素化;如果靶蛋白的多个赖氨酸残基同时被单一泛素分子标记称为多泛素化;而能够对单泛素化的靶蛋白进行泛素链的延伸则称为多聚泛素化。其中,有关第48号赖氨酸(K48)和第63位氨基酸(K63)位点多聚泛素化的研究最为广泛。蛋白质的多聚泛素链发生在泛素的K48主要与蛋白质降解有关;若发生单泛素化或在泛素的K63形成多聚链则主要参与蛋白质的内吞、转运等活动[12]。
1.2 E3泛素连接酶E3泛素连接酶,又称泛素连接酶,是一类能够将泛素分子连接到底物蛋白的某个赖氨酸上的酶,在泛素化的一系列酶促级联反应当中,它们可以特异性识别底物蛋白,还介导了泛素分子由E2s转移到底物蛋白的整个过程,在泛素化系统活性的调控中起着最重要的作用[7]。E3泛素连接酶参与泛素化底物蛋白的作用机制见图1。人体内表达600余种E3泛素连接酶,它们以特定规则结合不同底物蛋白,从而参与其泛素化过程[13]。
根据这些E3泛素连接酶的分子结构和作用机制,其可分为HECT(homologous to E6-associated protein 1 carboxy terminus)结构域家族、RING(really interesting new gene)结构域家族和U-box结构域家族3大亚型[14]。其中,HECT家族的E3泛素连接酶利用保守的半胱氨酸接受由E2s转移来的泛素分子,再将其转移到底物蛋白上,而含有RING和U-box结构域的E3泛素连接酶都可以与E2s相互作用从而将泛素直接从E2s转移到底物蛋白将其泛素化修饰。这种酶促性质以及其特异性底物识别的功能使得E3泛素连接酶能够成为AD潜在的治疗靶点。
2 E3泛素连接酶在AD中的作用
2.1 E3泛素连接酶通过参与泛素化APP减少Aβ的产生AD患者脑中由于Aβ的异常聚集而形成脑内淀粉样斑块,研究显示,在AD患者脑中,当未折叠或错误折叠的APP蛋白增多时,最后由UPS降解这些蛋白,从而减少Aβ的产生,进而减少AD患者脑内淀粉样斑块的沉积,所以E3泛素连接酶同样也是APP泛素化过程中最为关键的酶。
3-羟基-3-甲基戊二酰还原降解酶(Hrd1p/Der3p,HRD1)是一种E3泛素连接酶,其定位于内质网中。内质网在新膜结构的合成、分泌蛋白的加工、糖基化和二硫键的形成过程中起着功能性作用[15]。如果体内出现氧化应激或钙离子稳态失衡的情况,内质网中错误折叠或未折叠的蛋白质就会增多,进而引发内质网相关降解途径(ER-associated degradation,ERAD),最终由UPS降解清除这些蛋白质,而E3泛素连接酶HRD1可以促进因内质网应激产生的大量未折叠蛋白的降解。越来越多的证据表明,HRD1在脑神经元内质网的质量控制中起着重要作用。有研究发现在AD 患者大脑皮层中 HRD1的蛋白水平显著降低,且HRD1可以促进 APP 的泛素化和降解,使Aβ 的产生减少,而 HRD1的缺失会导致APP 积累和 Aβ 生成增加,加速AD疾病进展[15]。F-box和富含亮氨酸重复蛋白2(F-box and leucine rich repeat protein 2,FBL2)是Skp1-Cullin1-F-box蛋白(SCF)E3泛素连接酶复合物的一个组成部分,可通过自身F-box结构域和富含亮氨酸的重复区参与对底物蛋白泛素化的调控,在AD患者大脑中FBL2表达水平下降。FBL2能够与APP的羧基端特异性结合促进APP泛素化,进而调节APP代谢,降低神经细胞中Aβ的分泌量[16]。此外,由于APP从质膜内吞进入内体,然后被β-分泌酶(β-site amyloid precursor protein cleaving enzyme 1,BACE1)和γ-分泌酶切割是Aβ产生的主要途径之一,而FBL2可以通过抑制APP的内吞作用使脂筏中APP蛋白含量降低,增加了质膜表面APP的数量,减少BACE1裂解APP,可减少Aβ的产生。
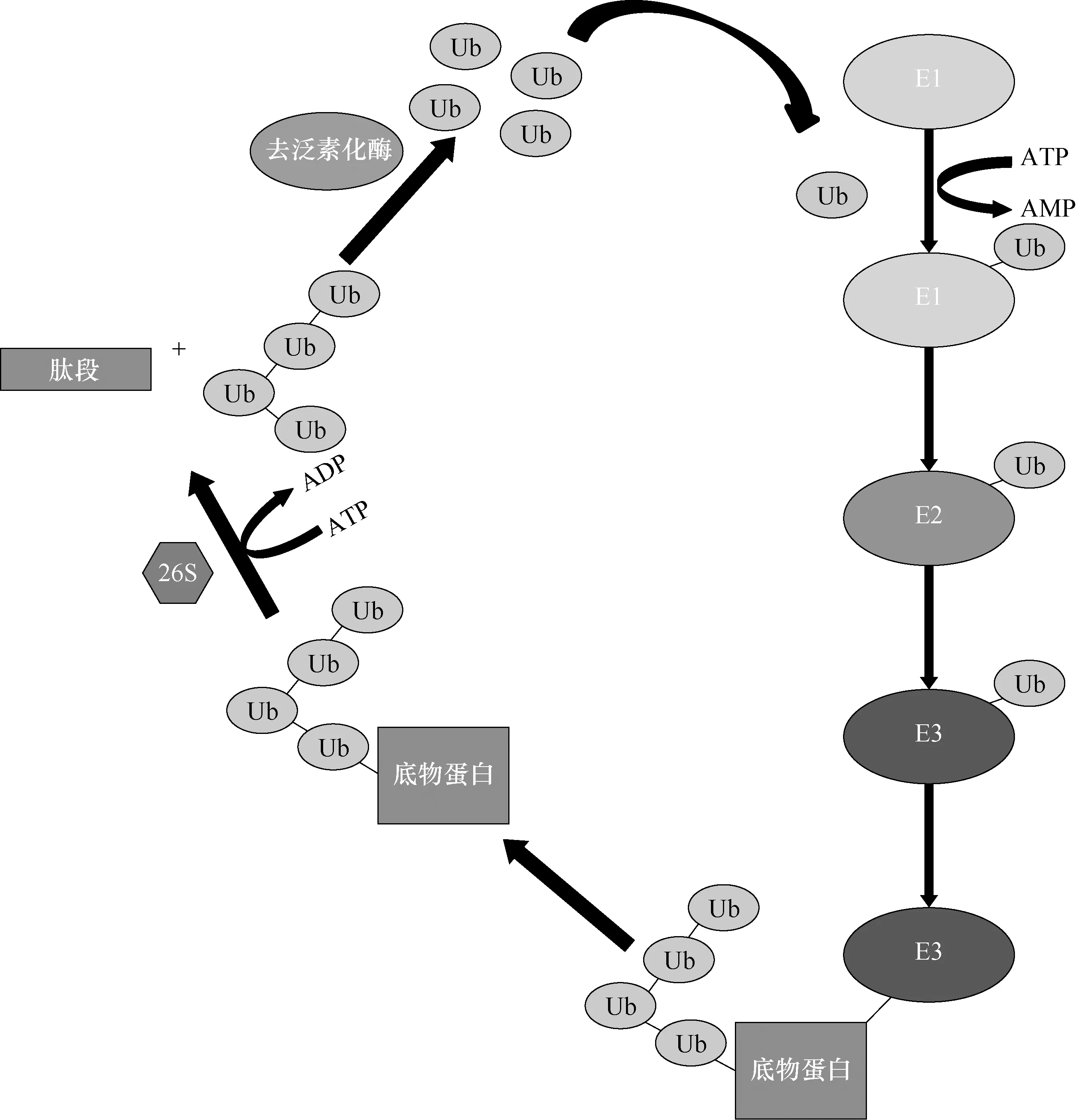
注:E1:泛素激活酶;E2:泛素结合酶;E3:E3泛素连接酶;Ub:泛素;26S:26S蛋白酶体 图1 E3泛素连接酶参与泛素化底物蛋白的作用机制示意图:首先,E1在ATP供能的条件下,其半胱氨酸残基与泛素结合并激活泛素分子。然后,活化的泛素通过硫酯键与E2的活性半胱氨酸位点结合。随后,E3泛素连接酶完成泛素从E2到底物蛋白的转移。最终,泛素标记的底物蛋白被26S蛋白酶体识别并降解。在这三种酶中,E3泛素连接酶是底物蛋白泛素化过程中最为关键的酶
有研究表明,AD1/FBL2转基因小鼠海马神经元中Aβ的蓄积明显减少[16],故增强 FBL2功能有望成为 AD 的一种新的治疗策略,可促进神经元内Aβ的减少。除HRD1和FBL2以外,还有其他E3泛素连接酶也具有相同的作用。热休克蛋白70羧基末端相互作用蛋白(carboxyl terminus of the Hsp70 interacting protein,CHIP)是一种无处不在的细胞质蛋白,在多个物种中结构保守,在大脑、心脏和肌肉中高度表达。CHIP的N端有三个四肽重复区,它们可与热休克蛋白(heat shock proteins,HSPs)70/90和其他分子伴侣蛋白,包括 Hip、Hop、Cyclophilin 40、FKBP52和Bag1磷酸酶5相互作用。CHIP的C末端包含一个RING结构域,这是其E3泛素连接酶的活性位点,故CHIP可以通过重新折叠和降解错误折叠的蛋白质来控制蛋白质的代谢。在AD患者脑中,CHIP参与了APP以及Aβ的降解。CHIP的过度表达一方面可以通过改变APP蛋白的成熟速率来稳定其表达水平,并诱导APP蛋白的正确折叠,另一方面,CHIP也促进APP与泛素分子的结合,为其进一步降解做准备。此外,CHIP可与多个HSPs结合形成复合物,降低SH-SY5Y 神经母细胞瘤细胞和皮层原代神经元Aβ42肽的水平[17]。
衰老是导致AD的高危风险因素之一。65岁以后患 AD 的风险每5年增加1倍。衰老可以加速E3泛素连接酶Mahogunin(MGRN1)的损失[18]。MGRN1在细胞和神经元保护中发挥重要作用:即它可以对抗胞浆蛋白和多聚谷氨酰胺神经毒性聚集体,保护神经元免受氧化和内质网应激的损害[19]。此外,MGRN1表达可以延迟APP的成熟。正常情况下,MGRN1将APP隔离在高尔基体,使得转运到细胞表面或进入内体/溶酶体室被BACE1裂解的APP减少,延缓APP的加工过程,最终减少Aβ 肽释放到细胞外[18]。因此,神经细胞培养中降低 MGRN1水平会导致 Aβ 肽释放增加。
以上研究均表明E3泛素连接酶通过抑制APP的淀粉样裂解途径,从而减少Aβ相关的神经元功能障碍,是APP加工和转运的强有力的调节因子。
2.2 E3泛素连接酶参与调节神经元凋亡和突触功能障碍神经元凋亡和突触功能障碍是AD的突出特征。一些E3泛素连接酶功能异常可以导致相关蛋白的稳态失调,进而影响突触结构功能,以及神经元稳态和信号传递功能,促进神经元凋亡。
突触功能障碍是AD记忆损害的原因之一[20]。支架蛋白 PSD-95定位于突触后致密区( post-synaptic densities,PSDs),在调节兴奋性突触信号传导中起重要作用[21]。PSD-95和相关支架蛋白 PSD-93的缺失或失调会损害突触功能,从而影响学习和记忆[22]。已有报道,在人类 AD 患者和 AD 小鼠模型中PSD-95和 PSD-93水平降低[23-24],提示突触功能障碍是 PSD 支架蛋白成分稳态失调的结果。UPS可以调节PSD-93/PSD-95的代谢[25]。UPS 对于调节神经元内的蛋白质环境,特别是调节突触后致密区内环境至关重要[26]。E3泛素连接酶鼠双微体2(murine double minute 2,MDM2)可以介导 PSD-93和PSD-95的降解,UPS依赖性的PSD成分失调在 AD 发病机制中扮演重要角色。有研究表明,RPS23RG1是一种跨膜蛋白,通过与腺苷酸环化酶的相互作用,可同时抑制Aβ和tau 过度磷酸化的产生,且RPS23RG1可以通过减少MDM2依赖的PSD-93/PSD-95降解来使AD的突触功能失调恢复[27]。在AD等神经退行性疾病中,终末分化的神经元细胞周期受到抑制对于维持神经元稳态至关重要,其异常激活会导致细胞周期重返,从而导致神经元凋亡(cell cycle re-entry that causes neuronal apoptosis,CRNA)。Itch是神经前体细胞表达发育调控蛋白4(neural precursor cell expressed developmentally down-regulated 4,NEDD4)家族的 E3泛素连接酶[28],它主要通过促进神经元中TAp73的降解来实现对CRNA的调控。TAp73可以调节microRNA-34a的表达,microRNA-34a在神经元分化和神经突生长中起重要作用。在AD中,Aβ42可以通过异常激活c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)信号通路导致E3泛素连接酶Itch过度磷酸化,被异常修饰的Itch会进一步泛素化降解TAp73,使神经元细胞周期发生重返[29],重要的神经元细胞周期蛋白表达异常,最终导致神经元的凋亡,加快AD的疾病进展。
环指蛋白(ring finger protein,RNF)182是E3泛素连接酶RNF183家族中的成员之一,其最初被确定为AD患者死后大脑中的一个上调基因,通常在神经系统中选择性表达,如大脑皮层、海马、小脑和脊髓,在心脏、肝脏、肾脏和骨骼肌中不表达。RNF182在AD患者脑中表达增加。RNF182在分化的人多潜能畸胎瘤干细胞(ntera2,NT2)中表达,并且在缺氧和葡萄糖缺乏的情况下表达会显著升高。研究人员通过酵母双杂交筛选发现,V型质子ATP酶16kDa蛋白脂亚基(ATP6V0C)是RNF182的底物。ATP6V0C是缝隙连接和神经递质释放通道的一个关键组成部分,在SH-SY5Y神经母细胞瘤细胞中敲低ATP6V0C蛋白可诱导神经毒性标记物增多,缩短神经元突起的长度。而RNF182可以通过蛋白酶体降解途径促进ATP6V0C的降解,最终导致神经元凋亡。故抑制RNF182表达或使其失活有利于改善AD的疾病进展[30]。
此外,研究人员还发现泛素连接酶E3A(Ube3A/E6-AP18),其在调节突触功能和突出可塑性方面也有重要作用。Rho蛋白(Rho GTPase)家族的RhoA、Rac1和Cdc42可以调节神经元树突棘的结构和功能,RhoA促进树突棘的失稳,而Rac1和Cdc42则促进其稳定和成熟[31]。另一个与AD患者突触功能障碍有关的的重要分子是突触毒性蛋白Arc。Ube3A的底物包括Arc和RhoA特异性核鸟苷酸交换因子(GEF)Ephexin-5。有研究表明,Ube3A在Tg2576小鼠脑中表达随年龄增长而下降,而Ube3A减少会导致突触毒性蛋白Arc以及Ephexin-5的积累,分别诱导突触表面的谷氨酸受体减少以及活性RhoA的增加,最终导致突触功能异常[32]。此外,Ube3A还可以激活重组人雌激素受体β(ESR2)基因的转录过程。ESR2通过调节脑源性神经营养因子(BDNF)来维持突触可塑性。在AD大鼠模型中过量表达ESR2可以减少海马区的Aβ沉积,并改善AD大鼠的学习和记忆[33]。
2.3 E3泛素连接酶参与神经炎症和免疫功能的调节AD的另一重要病理特征是慢性神经炎症。神经炎症一般指中枢神经系统(central nervous system,CNS)内的可由各种病理损伤,包括感染、创伤、缺血和中毒引起的炎症反应。炎症过程的特点是产生促炎细胞因子〔包括白细胞介素(interleukin,IL)-1β、IL-6、IL-18和肿瘤坏死因子(tumor necrosis factor,TNF)〕、化学因子〔如趋化因子配体1(chemokine ligand 1,CCL1)和CCL5〕,以及CNS的先天免疫细胞产生的活性氧[34]。参与这一过程的先天免疫细胞主要是小胶质细胞和星形胶质细胞,还有神经元和内皮细胞,均能够启动炎症反应[35]。
小胶质细胞是CNS的组织驻留巨噬细胞,与神经元和其他早期胚胎阶段的神经胶质细胞发育密切相关,在CNS的发育和稳态中起着关键作用[36-37]。有研究表明,星形胶质细胞和小胶质细胞介导的慢性神经炎症是AD的病理特征之一。CCAAT/增强子结合蛋白(CCAAT/enhancer binding protein,c/EBP)家族的转录因子与神经退行性疾病和脑损伤中观察到的炎症有关。这些转录因子调节激活小胶质细胞的关键基因[38-39]。其中,c/EBPβ与人类AD进展直接相关。而E3泛素连接酶 COP1,也称RFWD2,一种多亚基cullin-RING泛素连接酶CRL4COP1/DET1的底物接合分子,是转录因子 c/EBPβ和小胶质细胞介导的神经炎症的重要负调节因子。研究人员发现,小胶质细胞特异性缺失COP1会导致AD模型小鼠的激活型小胶质细胞和星形胶质细胞显著增加,神经退行性病变和海马萎缩更为严重。因此COP1缺失能够导致c/EBPβ迅速累积,驱动促炎和神经退行性相关基因的表达,从而加快AD的神经退行性病变[40]。E3泛素连接酶Pellino 1(Peli1)在小鼠大脑中的多种神经细胞中表达,在小胶质细胞中表达水平最高[41],这意味着Peli1在CNS中也具有非常重要的功能,以调节小胶质细胞相关的神经疾病。已有研究表明Peli1是小胶质细胞介导的自身免疫性神经炎症和病毒性脑炎的关键调节因子[42]。在小胶质细胞中,c/EBPβ是负责清道夫受体CD36基因转录的主要转录因子,而Peli1可以直接靶向c/EBPβ并介导其泛素化降解过程。因此,Peli1的缺失就会增加C/EBPβ的蛋白水平和CD36的表达,从而促进了小胶质细胞的吞噬能力,加速了AD模型小鼠脑内Aβ的清除[43]。故E3泛素连接酶Peli1可能是AD等神经退行性疾病的新的关键治疗靶点。肿瘤坏死因子受体相关因子(TRAF)是TNF超家族的重要结合蛋白,在天然免疫和获得性免疫中起重要作用。其中E3泛素连接酶TRAF6与AD等神经系统疾病密切相关,其可以通过toll样受体4(TLR4)/TRAF6/IκB激酶(IKK)/核因子-κB(NF-κB)信号通路介导炎症反应[44]。AD患者脑中的Aβ斑块周围的星形胶质细胞和小胶质细胞因Aβ与TLR-4相互作用而被激活,这种相互作用导致髓样分化因子88(MyD88)和TRAF6的激活,然后刺激NF-κB、激活蛋白-1(AP-1)和丝裂原活化蛋白激酶(MAPK)信号,导致促炎症细胞因子的释放[45]。促炎性细胞因子如IL-1β和TNFα以及活性氧均具有神经毒性[46]。
2.4 E3泛素连接酶影响线粒体功能线粒体在真核细胞中广泛分布,为细胞能量产生的关键细胞器,同时也参与氧化应激、细胞凋亡等多项细胞内的重要生理过程。因此,线粒体对于神经元正常的生理功能具有重要意义。近年来,大量研究表明线粒体功能障碍在AD的发病中发挥着非常重要的作用,AD早期即有线粒体功能障碍的表现。而AD患者脑内一些E3泛素连接酶,包括Parkin、四肽重复结构域3(tetratricopeptide repeat domain 3,TTC3)等功能异常或缺失均会加重线粒体功能的损伤。
PTEN诱导激酶1(PTEN-induced kinase 1,Pink1)和 Parkin RBR E3泛素连接酶可以调控功能失调和过剩的线粒体的特异性清除,从而使线粒体网络结构和能量代谢保持稳定。Pink1在受损的线粒体中调节Parkin易位,并通过选择性自噬驱动它们被清除,此过程被称为线粒体自噬[47]。Parkin的E3泛素连接酶活性可以介导线粒体蛋白泛素化促进线粒体自噬。因此Parkin缺失会导致线粒体功能障碍,加剧炎症并促进小胶质细胞活化,促进神经炎症。在AD疾病进展过程中,胞浆Parkin水平降低,使得线粒体功能障碍增加[48],最终导致突触和神经元受损,认知功能障碍加重。线粒体功能受损不仅影响线粒体功能过程,而且导致蛋白进入线粒体的阻滞,从而引起线粒体前体蛋白在胞浆内积累,导致蛋白失衡。DNA聚合酶γ(POLG)是DNA聚合酶的催化亚单位,是位于线粒体中参与线粒体DNA复制和修复的酶,对于维持细胞功能和完整性至关重要[49]。因此,当POLG发生突变时,线粒体功能受到损害,影响大脑等耗能器官[50]。E3泛素连接酶TTC3可以调节POLG进而影响线粒体功能,严重的线粒体损伤会引发线粒体自噬。有研究表明,可溶性的TTC3会促进POLG的降解,而高水平的TTC3过表达会导致聚集物形成并阻止POLG的降解,因此,POLG的稳定性在两个方面受到E3泛素连接酶TTC3的影响,均可以导致POLG功能的丧失[51],最终影响AD患者的认知功能。
综上,AD 是一种多病因参与的复杂疾病,多种E3泛素连接酶的缺失和异常是其中的一个重要致病因素。E3泛素连接酶能够介导APP的泛素化降解及转运,最终影响Aβ的生成,能够介导神经元和突触相关蛋白质的泛素化降解,还能直接促进泛素化转录因子,从而抑制小胶质细胞介导的神经炎症及其吞噬能力,也可以通过影响线粒体功能进而影响AD的发生发展。然而UPS作用机制十分复杂,是否有新的E3泛素连接酶可以调控AD关键致病分子,以及其是否可以作为新的AD临床治疗靶点,值得我们继续深入探讨。因此,应加深对UPS、E3泛素连接酶以及其在AD的疾病机制中所起到的作用的了解,以期为后续治疗AD等神经退行性疾病的药物研究开发提供新的思路和新的靶点。
