深度学习在药物活性预测研究中的应用*
2022-08-20刘利梅陈晓晋孙世伟梅树立王耀君
刘利梅 陈晓晋 孙世伟 王 宇 王 辉 梅树立** 王耀君**
(1)中国农业大学信息与电气工程学院,北京 100083;2)中国科学院计算技术研究所,北京 100190)
药物研发是一项周期长、风险高、投资大的产业。分子类药物研发过程一般分为5个阶段:药物新化合物制备、药物临床前研究、药物临床实验、药品申报审批和新药监测。药物新化合物制备过程主要涉及到药物化学的应用。药物化学是一门结合了高分子化学和生命科学的综合性学科,该领域的研究可以分为3类,分别是药物分子的探求、药物分子的修饰和药物分子的结构调整。其中药物分子的探求耗时最长,风险也最大,需要消耗更多的人力和物力。在药物分子探求过程中,药物活性预测能够协助筛选出具有活性的化合物,从而作为药物的先导物参与后续药物研发进程,提高药物研发的成功率和效率。
药物活性是指分子药物的生物活性,是确认化合物能否作为药物先导物的主要指标。药物活性预测是指分析候选药物分子与生物体中靶标蛋白的结合效果,即针对某种疾病的潜在疗效。在药物筛选的过程中通常根据预测结果对候选分子的活性进行打分和排名,选择疗效最优的药物分子作为药物的先导物。随着药物研发技术的逐步发展,尤其是在近期新型冠状病毒疫情蔓延时期,药物合成和研发愈发受到重视。通过借助生物化学技术的快速发展以及与新型科技的结合,目前每天都会有成千上万种新型化合物分子被人工合成,其中包含不少用于治疗各类疾病的药物分子。药物分子发现和分子合成是当前新药研发的重要途径,及时准确的药物活性预测会大大加快新药研发的进程。
以机器学习为代表的人工智能算法可以辅助解决药物研发中药物活性预测耗时较长、准确度低、效率低下等问题。在药物研发中,机器学习算法主要应用于药物分子设计、药物分子与靶标蛋白的相互作用分析、药物的吸收分析、药物代谢分析、药物毒性分析等方面。机器学习能够通过对某一类问题的数据特征进行学习,进而对某一事物或者问题进行分类或者回归预测。
深度学习是机器学习研究领域的一个热门研究分支,是人工神经网络的进一步拓展。传统机器学习方法的应用使药物活性预测效率和准确度有所提升,但仍然难以满足当前精准医疗和精准医药的需求。以深度学习为代表的神经网络模型通过借鉴人脑的多分层结构、神经元信息交互的逐层分析处理机制,能够自适应、自学习地对信息进行并行处理,在生物医药的较多领域取得了突破性进展。目前,不同的深度学习网络模型在药物活性预测方面取得了较好的成果。深度学习领域新方法新模型的层出不穷大大提高了药物活性预测能力并推动了药物研发领域的不断发展。
本文对近年来药物活性预测方面的研究和应用进行了综述和分析,重点分析了深度学习在药物活性预测方面的研究。文章整体结构按照数据、算法、应用、总结的方式进行构建,具体的应用实例按照数据、方法、结果的顺序进行分析。药物活性预测的分类及方法流程如图1所示,方法主要分为两大类,以实验为主的方法和以计算为主的方法,以计算为主的方法又分为统计分析、传统机器学习和深度学习三类。在药物活性预测环节,无论采用什么方法,其总体目标都为得到药物活性最高的先导化合物。图2为药物活性预测的方法和资源轮廓图,汇总了本文所涉及的数据库以及药物活性预测的传统方法、机器学习方法和深度学习方法及其对应的文献信息。
文章接下来的内容分为以下几个部分:第1部分“数据库及数据类型”,总结分析了当前应用于药物研发的重要数据库;第2部分“传统药物活性预测方法”,总结分析了药物活性预测的传统方法,这里的“传统”指的是方法中涉及到计算的环节未采用机器学习类算法;第3部分“传统机器学习应用于药物活性预测”,从算法原理到实际应用分析总结了传统机器学习算法在药物活性预测中的应用,这里的“传统”指的是在方法中未采用当前机器学习领域中热门的深度学习类算法;第4部分“深度学习应用于药物活性预测”,从算法原理到实际应用分析总结了深度学习算法在药物活性预测中的应用;第5部分“总结”,对文章前面几个部分的内容做了总结;第6部分“展望”,提出了药物活性预测领域的下一步可能的发展路径,同时提出供本领域的研究者或即将开展本领域研究的相关研究者参考的研究方向。
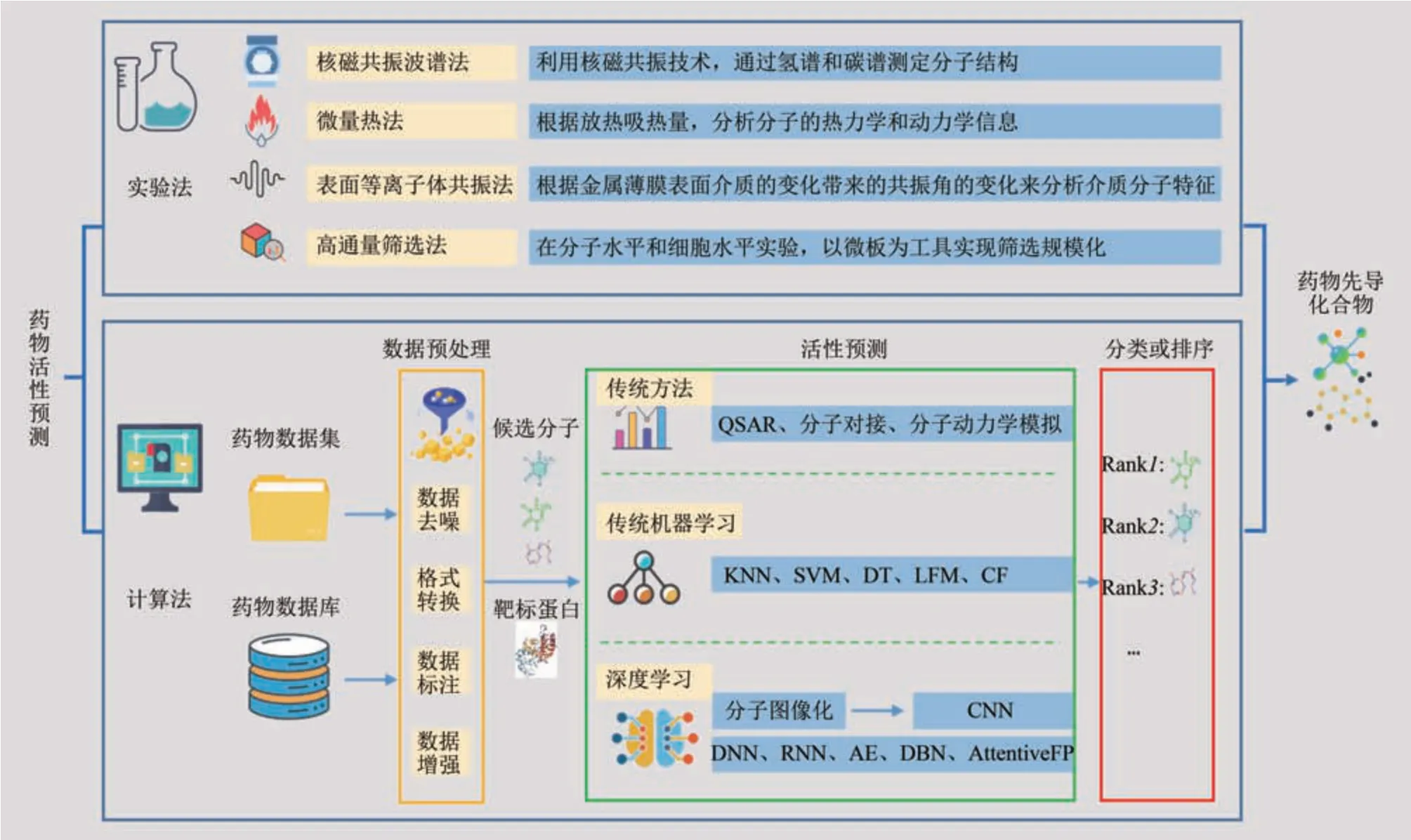
Fig.1 Flow chart of drug activity prediction图1 药物活性预测流程图

Fig.2 Summary of drug activity prediction databases and methods图2 药物活性预测数据库及方法汇总
1 数据库及数据类型
从计算的角度进行药物活性预测,本质是对药物活性预测问题的计算机建模。而建模的基础是支撑模型构建和运行的基础数据。同时,模型的有效性和准确性很大程度上取决于数据的数量和质量,所以药物研发相关的数据集及数据库的获取是药物活性预测建模的基石。本部分内容首先介绍了药物活性预测相关的主要数据库资源;接着分析其主要数据结构和数据类型;最后重点分析几种常用的药物靶标特征表示方法。
1.1 药物活性预测相关数据库资源
当前应用于药物活性预测的相关数据库资源主要 有:DrugBank[1]、ZINC[2]、ChEMBL[3]、PubChem[4]、KEGG BRITE[5]、PDBbind[6]、STITCH[7]、BindingDB[8]和PharmMapper数 据库[9]等。
DrugBank是一个服务于药物研发,包含有生物信息学和化学信息学数据的数据库,由加拿大Alberta大学的研究人员创立和维护;该数据库提供超过50 000种药物及其衍生物的化学结构、药理、药物作用以及靶标蛋白等较为全面的数据。ZINC[2]是由加州大学的Irwin和Shoichet在2005年创建的用于虚拟筛选市售化合物的数据库。截至2021年10月,数据库收录了7.5亿条包含有注释信息的小分子化合物数据,注释信息包含分子质量、LogP模型值,以及可应用于分子对接的结构数据。ChEMBL数据库由欧洲生物信息研究所开发,是一个能够大规模提供化合物生物活性及靶标蛋白数据的数据库。截至2021年7月,该数据库收录了14 554种靶标蛋白、2 105 460种化合物、18 635 916种活性化合物相关信息以及81 544份公开发表文章,并提供易用的生物活性数据的查询工具。
PubChem数据库是世界上最大的开源化合物数据库。截至2021年10月,该数据库收录了110 040 027条不同化合物的分子结构、生物检测、生物活性数据、基因序列、靶标蛋白、关联文献、关联专利等信息,同时还收录了独立实验室上传的生化实验数据及化合物数据等,目前该数据库数据依然保持持续更新。KEGG BRITE数据库包含基因序列、蛋白质、化合物反应、药物、器官和细胞等各种不同层次生物对象的数据,数据以BRITE结构层次文件表示。PDBbind数据库创建的目的是完整收录PDB数据库中通过实验测定的亲合力数据,数据库提供了生物分子复合物的亲和力及分子结构信息。数据库自2004年创建以来,保持每年更新一次,截至2021年10月,该数据库收录了23 496个生物分子复合物的数据。STITCH数据库和BindingDB数据库都录入了各类小分子化合物与不同靶标蛋白之间的相互作用关系数据。PharmMapper是由刘晓峰等[9]开发的用于药效团匹配与识别潜在靶标信息的数据库平台,该平台以活性小分子为探针搜寻潜在药物靶点,进而对小分子药物活性进行预测。平台中内嵌的统计方法可以自动查找与分子构象最为匹配的药效团并根据匹配度打分进行排序。
通常,在药物研发的药物分子设计中,药物靶标信息需要使用多个数据库的联合查询和汇总。例如,在药物研发中,对DrugBank的XML文件进行解析可以获得相关药物信息之外的ChEMBL ID、PubChem Compound ID等,通过这些ID可以将不同的数据库联合起来分析靶标蛋白、小分子药物相似度关系以及进行药物毒副作用关联。
1.2 数据结构及类型
药物研发相关数据库支持对药物分子化学结构、药理、药物作用以及靶标蛋白等数据的下载。但是,不同的数据库导出信息往往包含有不同类别的信息表示方式及文件存储格式,存在数据多源、结构多元等特点。在具体数据分析及建模环节需要根据文件格式编写相应的数据读取及格式转换接口程序。本研究对药物设计领域最常用6个数据库的数据结构和文件格式进行了详细分析,分别是:DrugBank、ZINC、ChEMBL、PubChem、KEGG BRITE和PDBbind,下面分别进行分析。
a.DrugBank支持下载数据库汇总的所有药物信息,不同的信息类别对应不同的数据结构及文件格式。其中,药物的完整数据对应的是XML文件,结构体信息是SDF文件、外部链接的数据和蛋白质标识符信息是CSV文件、目标蛋白质序列信息是FASTA文件。SDF文件中的第一行字符一般为分子名字,第二行是对分子的注释说明,第三行是空行,从第四行开始记录结构的原子数、化学键的个数等信息,直到出现“MEND”标识符,表明原子和化学键信息记录的结束,后面数据会保存多行关于分子属性的信息,以4个美元符号作为SDF文件结尾标识符。
b.ZINC数据库支持下载SMI、2D、3D和简化的分子输入行输入系统(simplified molecule input line entry system,SMILES)格式的文件。
c.ChEMBL支持下载采用SMILES化学结构表示法的分子结构SMI文件和包含Canonical SMILES表示法及InChl表示法的txt文件。
d.PubChem对于药物化合物的数据及药物关联信息数据,有CSV、JSON、XML 3种可选数据导出文件格式。对于药物分子结构信息有SDF、JSON、XML、ASNT、PNG 5种可选数据导出文件格式。
e.KEGG BRITE数据库的数据结构为keg格式的文本文件。文件分别以A、B、C、D为行开头的标识符来标记不同类别的信息;C开头数据行记录KEGG的药物pathway的ID,其药物pathway通常包含代谢、通路、调控、生化等相关的分子相互作用信息;D开头的数据记录相关联的基因组信息;A、B是KEGG数据分类标准的分类信息。
f.PDBbind支持下载用于药物分子建模的蛋白质-配体解离动力学参数数据集。数据集采用PDB格式保存蛋白质数据,以Mol2和SDF格式保存配体数据。
通过对现有药物研发相关数据库的数据结构及数据文件格式分析,可得出如下结论:SDF文件常用来保存化学分子结构;FASTA文件常用来存储蛋白质氨基酸序列信息;SMILES和InChl是常用的化学结构线性表示方式;JSON和XML格式的文件是生物信息领域最受青睐的数据组织格式,易于计算机程序分析和数据解析。
1.3 药物靶标特征表示方法
药物活性预测的目标是筛选与药物靶标蛋白结合效果最优的先导化合物分子。药物靶标是指生物体内具有药效功能并能被药物作用的生物大分子,即可药化大分子,例如,可药化的蛋白质、核酸等。靶标蛋白也称作可药物化蛋白质,是指可以调节与药物相互作用并且利用它们之间的相互作用产生治疗效果的蛋白质。
在药物研发过程中,需要对药物分子和靶标蛋白之间的关系进行数量化特征表示,用于进一步建模。常用的分子特征表示有:分子描述符、相似度矩阵和神经网络指纹[10],下面分别对其特点进行分析。
分子描述符(molecular descriptors)[11-12]方法分为定量描述符和定性描述符。该方法的原理是将分子结构内编码的化学信息转换为数值矩阵。分子描述符包括:分子结构、理化性质、分子图论、分子光谱数据、分子场以及分子形状的描述符信息;定性描述符又称分子指纹,可以用某种数据编码来表示分子的结构、片段、性质或子结构,常用的分子指纹包括:MACCS Keys[13]、ECFPs(extendedconnectivity fingerprints)[14]、Public Keys[15]、Daylight Fingerprint[16]等。根据描述符计算所需的信息维数,分子描述符可分为1D、2D和3D描述符。其中1D描述符表示分子的化学成分;2D描述符增加了原子之间的连接信息;3D描述符进一步增加了描述药物分子的3D坐标和绝对参考系。
药物的分子描述符除了用于药物活性预测,还可以结合不同的机器学习分类器对未知结构的蛋白质进行热点残基预测[17]。分子描述符方法相关的软 件 及 软 件 包 有:Rcpi[18]、PROFEAT[19]、iFeature[20]、Padel-Descriptor[21]、DRAGON[22]、CDK[23]、CODESSAPro[24]和MOE[25]等。
相似度矩阵表示法一般采用分子描述符和蛋白质序列比对匹配度评分来分别计算分子药物相似性和靶标蛋白相似性,然后以数值矩阵的形式存放其相似值。该方法目前常用于核回归[26]、二分局部法[27]和矩阵分解等模型。相似度矩阵表示方法简单易用,但是丢失了化合物的结构信息,无法处理未知的药物和蛋白质数据。
神经网络指纹的思想由Merkwirth等[10]提出。神经网络指纹表示法采用了类似于词向量模型Word2vec[28]的思想,借助深度神经网络模型将化合物分子离散的化学结构映射到连续的向量空间中,进而使用连续优化方法对模型进行优化。神经网络指纹不需要任何预定义的分子描述符,其在网络模型的训练过程中能够自动生成特征向量。另外,神经网络能够自适应地针对不同任务不同数据集学习到不同的指纹特征,所以能够获得更高的预测精度。与传统的表示方法相比,神经网络可以根据隐层的节点数确定分子特征向量的长度[10]。
2 传统药物活性预测方法
药物活性预测之所以是药物研发的必要环节,是因为药物进行疾病治疗的过程,通常也是药物分子与对应的靶标大分子相结合的过程。通过改变靶标分子的生理活性进而实现对疾病的治疗。药物和靶标之间相互作用关系的准确识别是推动药物研发效率提升的基础。早期的“一药一靶一病”理论没有考虑药物和靶标蛋白之间复杂的相互作用关系。当前的“多靶标-多蛋白”模型和药物重定位理论由英国Dundee大学药理学家Hopkins[29]于2007年提出,揭示了药物和靶标蛋白质复杂的相互作用,即某种药物的靶标蛋白可能会和其他药物进行结合,某一特定药物也可能会和其他蛋白质进行结合,造成脱靶效应[30]。
目前,药物靶标蛋白相互作用研究主要分为实验类的方法和基于计算类的方法。实验类方法主要有核磁共振波谱法(nuclear magnetic resonance,NMR)[31]、微量热法(microcalorimetry)[32]、表面等离子体共振法(surface plasmon resonance,SPR)[33]和高通量筛选(high throughput screening,HTS)[34]等。这些方法成本高、耗时长、应用范围小,故基于计算的药物靶标预测方法越来越受到药物研究人员的青睐和重视。传统的基于计算的预测方法主要有:基于配体的预测方法、基于结构的预测方法以及分子动力学预测方法(表1)。
2.1 基于配体的预测方法
在生物医药领域中,配体是指能够识别药物靶标并与之结合的分子。基于配体的预测方法主要是根据药物分子的三维结构进行活性预测,通常使用由Hansch等[35]提出的定量构效关系(quantitative wtructure-activity relationship,QSAR)来进行活性预测。该方法认为药物分子的活性与其结构具有直接相关性,可以通过建立相应的数学模型对药物分子结构和它的某种生物活性之间的对应关系进行表示。QSAR在药物化学领域具有较大影响力,但是该方法仍然具有一定的局限性,即如果与靶标蛋白一致的配体数量较少,则很难找出配体之间的相似特性来进一步预测药物的活性。
2.2 基于结构的预测方法
基于结构的预测方法主要通过对靶标蛋白三维结构进行分析进行活性预测。该类方法在分子对接(molecule docking)[36]上应用较为广泛。分子对接采用经验评分函数来对药物和靶标之间的相互作用进行评估。Wang等[37]提出了蛋白质-配体打分方法SCORE,该方法引入了原子结合评分,采用经验评分函数对结合自由能进行表示,进而对已知三维结构的蛋白质与相应配体的结合亲和力进行计算。其他经验评分方法还有基于力场的DOCK[38]和GOLD[39]、基 于 知 识 的DrugScore[40]和DFIRE[41],以及基于统计方法的X-Score[42]和VALIDATE[43]等。
2.3 分子动力学预测方法
分子动力学模拟(molecular dynamics simulations)是利用计算机模拟的方法对原子的物理运动以及多原子体系中分子的变化进行研究。分子动力学模拟在药物活性研究中能够实现受体构象搜索和小分子最佳结合位点选择。此外,可以通过结合自由能来评估药物分子和靶标蛋白之间的结合力的强弱。Miao等[44]采用加速分子动力学结合Glide诱导拟合对接的方法,成功筛选出国家癌症研究所中38种化合物中与M2毒蕈碱乙酰胆碱受体(mAChR)结合亲和力≤30μm的12种化合物并成功确定正负变构调节剂。

Table 1 Traditional drug activity prediction methods表1 传统药物活性预测方法
3 传统机器学习应用于药物活性预测
机器学习(machine learning,ML)是计算机通过与相关数学理论如统计学、概率论、逼近理论和复杂算法知识等进行结合,通过模拟人类的学习方式对经验进行学习,该种方法能够对现有的事物进行分类、预测或者决策。受生物实验方法低精度和高费用的限制,药物活性预测效率低下、准确率低、实际应用代价高。采用机器学习和深度学习算法进行药物活性预测能够在一定程度上缩短药物筛选的时间、降低研发成本、减少新药研制的盲目性,这对于医药行业的发展具有重要推动意义。
针对不同的药物活性相关数据资源及研究问题,可以采用不同的机器学习模式,机器学习模式主要分为:有监督学习、无监督学习和半监督学习。在有监督的学习模式中,训练样本全部为已知标注特性的实例数据,多用于对样本进行分类和回归;在无监督的学习模式中,训练样本没有经过标注,由算法根据特征进行自动分类和聚类,多用于样本聚类、异常值检测和降维;在半监督的学习模式中,训练样本中含有部分已知特性的标注样本和部分未知特性的样本,该模式通过增加未知样本来提高机器学习的自主性,当未标记数据阐明的实例分布与分类问题相关时,未标记数据可以帮助提高预测精度和加快学习速度[45],半监督学习模型多用于样本分类和聚类。
在药物活性预测相关研究中,具有代表性的半监督学习模型有LapRLS[46]和NetLapRLS[47],具有代表性的无监督学习模型有MG-BERT[48]和Mol2vec[49]。针对已知的药物-蛋白质相互作用的稀缺性和未知药物和蛋白质的相互作用有待预测的问题,LapRLS和NetLapRLS均采用半监督的学习方法对未知药物和蛋白质的相互作用进行预测。这两种半监督的方法在Nuclear Receptor、GPCR、Ion channel、Enzyme 4个开源数据集上的实验结果显示,综合利用有标记和无标记的数据,通常比单独使用有标记数据能产生更好的结果。
MG-BERT使用了无监督原子表达学习来进行分子的性质预测。模型的作者提出了分子图BERT结构,将图神经网络(GNNs)的局部消息传递机制集成到BERT模型中,以便从分子图中进行学习。该方法使用了一种有效的自监督学习策略,即掩蔽原子预测,对模型进行预处理,以挖掘分子中的上下文信息。在预处理后生成上下文敏感的原子表示,并将学习到的知识用于各种分子性质的预测。实验结果表明,在11个ADMET数据集上,经过微调处理的MG-BERT预训练模型的性能始终优于现有的同类模型。Mol2vec采用了一种无监督机器学习方法,用于学习药物分子结构的向量表示。类似于自然语言处理研究领域中的Word2vec模型,密切相关的词向量在向量空间中非常接近。Mol2vec模型能够对分子结构进行向量表示,相似的分子结构向量在向量空间中也非常接近。其输出的结果向量可作为输入变量,联合有监督的机器学习方法进一步预测化合物的性质。
3.1 常见机器学习算法原理简介
目前,常见的机器学习算法有多元线性回归(multiple linear regression,MLR)、朴素贝叶斯(naive Bayesian,NBM)、K‑近 邻(K-nearest neighbor,KNN)、K-means、随机森林(random forest,RF)、决策树(decision tree,DT)、套索回归(lasso regression,LR)、支持向量机(support vector machine,SVM)、Boosting与Bagging算法、逻辑回归(logical regression,LR)、岭回归(ridge regression)、人工神经网络(artificial neural networks,ANN)和深度学习算法(deep learning,DL)等。在药物活性预测方面,主要用到机器学习的分类功能。目前,比较常用的算法包括SVM、KNN、ANN、RF、深度学习等。
KNN是一类有监督的分类模型,其分类器无需训练并且可以解决多分类问题。该算法的原理为:对于给定测试样本,通过计算样本间距离寻找训练集中与该样本最靠近的K个样本,然后根据这K个样本的标注类别采用“投票法”对类别进行打分,K个样本中出现次数最多的类别即为输入样本的预测类别。KNN算法有3个关键要素:K值的选择、距离度量和分类决策规则。
DT是一类有监督的分类模型。通过训练数据构建决策树,然后对未知数据进行分类。该模型的树形结构中的每一个节点都表示一个属性,输入的待预测样本自上而下遍历树形结构并根据其属性完成多次判断,直到给出样本的最终判定结果。模型训练过程中主要特征度量方法有3种:信息增益、信息增益率和基尼指数[50],相应的算法分别为ID3、C4.5和CART(classification and regression tree)算法[50]。RF是对多个决策树的集成,通过投票表决输出样本类别。由于RF中每棵树的特征选择不同,因此可以解决过拟合问题,增强模型的泛化能力。
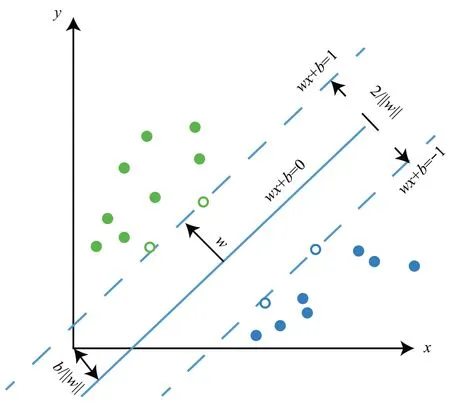
Fig.3 Support vector and interval图3 支持向量与间隔
SVM是一种有监督的分类模型,其在二分类问题上表现较好,针对一对多和多对多的分类问题效果略差。SVM的原理为:在训练集样本空间中寻找一个超平面,使得不同类别的样本分布在超平面的不同侧,该超平面可表示为图3中的多元线性方程。其中,w为超平面的法向量,代表超平面的方向,b为位移,代表原点和超平面之间的距离。常见的SVM扩展算法有:半监督支持向量机(semi-supervised support vector machine,S4VM)[51]、代价安全性半监督支持向量机(cost security semisupervised support vector machine,CS4VM)[52]等。
除了上述几种机器学习算法,基于矩阵分解的推荐算法如协同过滤(collaborative filtering,CF)[53]、隐语义模型(latent factor model,LFM)[54]以及基于网络的推荐方法[55]等在药物靶标活性预测上取得了较好的效果。CF算法是一种较为常用的推荐算法。在商品推荐领域,能够基于用户历史行为数据预测用户的喜好和偏向;在药物活性预测领域,通过基于邻域和统计学的非负矩阵分解算法,将“药物-靶标”之间的关联关系类比为商品推荐系统中的“用户-商品”场景。
3.2 机器学习算法在药物活性预测中的应用
在具体的行业应用中,机器学习算法需要借助大量有标注的和无标注的行业数据对模型参数进行学习和优化。在药物活性预测研究领域中,“类药五原则”理论(又称为Lipinski规则)[56]经常被用作活性化学药物的初筛。化学基因组学方法(chemogenomic approaches)[57]将药物化合物的化学空间和靶标蛋白的基因组空间进行整合,借助于强大的生物化学数据库资源,机器学习开始在化学基因组空间中展开应用[58](表2)。

Table 2 Traditional machine learning models applied to the prediction of drug activity表2 传统机器学习模型应用于药物活性预测
3.2.1DT及RF在药物活性预测中的应用
DT呈树形结构,在分类问题中,表示基于特征对实例进行分类的过程。国内学者贾聪敏[59]采用RF算法结合十折交叉验证构建药物靶点定量预测模型,所构建的RF模型在EC50验证集和测试集上的MSE均小于0.09,且R2均大于0.96;在KD数据集上MSE均小于0.12,R2均大于0.94。实验同时也设置了多种算法作为对照组,结果表明相比较于SVM和ANN,RF构建的药物靶点相互作用关系定量预测模型为最优模型。RF针对缺失数据和异常值不敏感,通过随机性的引入增强了模型的泛化能力,其在3种模型中表现最佳,ANN因在药物靶点定量预测上出现欠拟合问题而表现最差,SVM则介于两者之间。Lü等[60]采用C4.5 DT算法建立分类模型对肝炎病毒NS5B蛋白酶抑制剂与非抑制剂进行预测,在丙型肝炎病毒的基因复制和蛋白质成熟的过程中,NS5B蛋白酶是RNA复制依赖的聚合酶,抑制NS5B聚合酶可以阻止丙型肝炎病毒的RNA复制,因此成为一种治疗丙型肝炎的有效方法。该方法的测试数据集包含1 248个结构多样性的化合物(552个NS5B抑制剂与696个非NS5B抑制剂),实验结果表明,C4.5 DT算法在非抑制剂数据集上取得最高的预测精度(87.2%),在抑制剂数据集上取得最低的预测精度(81.4%),在总的数据集上,其预测精度(84.7%)略低于KNN(85.0%)。
3.2.2SVM在药物活性预测中的应用
SVM能够处理高维数据以及对线性和非线性数据进行分类。其在化合物分类、排名以及回归属性值预测方面具有良好的效果。在药物活性预测领域,通常被作为基础模型或对照组模型应用。
Lü等[60]测试了SVM在肝炎病毒NS5B蛋白酶抑制剂和非抑制剂上的分类预测模型,为了同时提高模型计算效率和预测精度,作者首先通过RFE(recursive feature elimination)对分子描述符中的分子特征进行筛选。模型的实现流程如下:a.针对数据集中的全部分子描述符建立模型;b.通过评分函数对分子描述符中的分子特征进行评分;c.删除排在最后的m个描述符;d.最后对剩下的描述符进行SVM分类训练,采用5折交叉验证计算模型的精度。
为避免模型的过拟合同时降低模型的计算量,设置变量m=5。在NS5B抑制剂和非抑制剂数据集上,使用RFE特征预筛选使得SVM的总预测精度从69.8%提升到82.0%。与KNN和C4.5 DT模型相比,SVM在抑制剂数据集上取得最高的预测精度91.7%,在非抑制剂数据集上取得最低的预测精度78.2%。由此可见模型的好坏与数据集有很大的关系。
高双印[61]分别对3类药物活性分子(PLK1 PBD、SMAD3、IL-1B)进行深入探究,分别基于SVM、S4VM和CS4VM构建药物活性预测模型,3种模型在PubChem里SMAD3数据集上的预测精度,分别为61.47%、75.16%和71.35%,其中S4VM取得了较好的预测效果。贾聪敏[59]采用SVM构建药物靶点EC50定量预测模型,其在训练集上的R2=0.931 7,MSE=0.127 0,在测试集上的R2=0.575 9,MSE=0.835 6,优于ANN模型,但相对于RF来说效果略差。
3.2.3推荐模型在药物活性预测中的应用
推荐模型主要用于广告、产品、电影推荐等应用场景。常用的推荐模型有矩阵分析和协同计算。在电子商务领域,推荐系统可以根据用户购买商品的历史数据和同类用户的购买数据进行建模然后进行个性化商品推荐。这一思想被研究人员应用于研究启发式药物发现,将药物和药物的关系以及药物和靶标的关系类比于广告推荐中的用户和用户关系以及用户和商品的关系。
何亚琼等[62]借鉴推荐系统对药物靶标预测问题进行建模,建模过程如下:a.建立表征药物靶标相互作用的m×n二维矩阵R,m代表药物量,n代表靶标数量。矩阵R中,如果已知药物i和靶标j存在相互作用,则Ri,j=1,未被验证的关系对应值为0;b.建立表征药物相似性的m×m的对角矩阵M,Mi,j是根据化合物i和化合物j的化学结构相似性计算得到;c.构建表征靶标蛋白相似性的n×n对角矩阵N。矩阵Ni,j是基于目标蛋白的氨基酸序列相似性计算得到;d.Ui和Vj分别代表药物i的潜在影响因子向量和靶标j的潜在影响因子向量,Kn,m是药物和靶标的潜在低维空间维度。
何亚琼等[62]提出了深度学习协同过滤算法,设计多输入深度自编码器,使其能够自动提取药物潜在特征U和药物靶标潜在特征V。通过在编码器中加入相似度矩阵M、N和药物靶标相互作用矩阵R,模型能够同时提取到这3种矩阵中的药物和靶标的潜在特征,进而增强U和V的有效性。另外,作者通过添加药物、靶标双重正则化来提高模型的精度。改进后的模型流程如图4所示。
与采用SVM做二分类的深度自编码器、基于低秩矩阵分解的协同过滤算法相比,在Yam基准数据集(Nuclear Receptor、GPCR、Ion channel、Enzyme)上该方法的AUPR值分别为0.764、0.732、0.682和0.637;在酶数据集上,AUC值达到了0.978,改进后的模型AUPR和AUC值均高于MFDR(SDAE+SVM)和COSINE。章啸[63]针对药物靶标关联矩阵过于稀疏的问题,采用混合加权方法对关联矩阵进行改进,并将DrugBank、ChEMBL、PubChem、Uniprot 4大数据库的数据融合为一个新的数据集,改进后的算法在该数据集上的MAE比直接使用协同过滤降低约0.1~0.15。
相对于早期协同过滤采用的奇异值分解方法,隐语义模型(latent factor model,LFM)是对奇异矩阵分解(singular value decomposition,SVD)的改进,同时LFM引入了损失函数和惩罚项来提高模型预测精度。Mongia等[64]提出一种深度隐语义模型,将矩阵分解推荐算法由分解成两个矩阵改为分解成多个矩阵。在Enzymes、Ion channels、GPCR和Nuclear receptors 4个基准数据集上,该模型的AUPR分别达到了0.728、0.828、0.616和0.125,AUC值分别为0.899、0.941、0.884和0.669,均高于其他矩阵分解方法如SVT、BMC、PMF和GRMF。
基于网络的推荐算法是指将网络接口接入不同的算法,使得用户可以根据自己的需求自定义训练数据,进而获得更加精准的预测结果。Salvatore等[55]应用基于双向网络投影的推荐技术提出了药物活性预测应用Web程序DT-Hybrid,用户可以通过系统上传待预测的药物数据,然后自定义设置各项参数,该系统通过DT-Hybrid返回一个预测的候选结果列表,并对每一个候选结果进行打分供用户参考,该系统的底层数据来自于DrugBank数据库,并且定期进行更新。
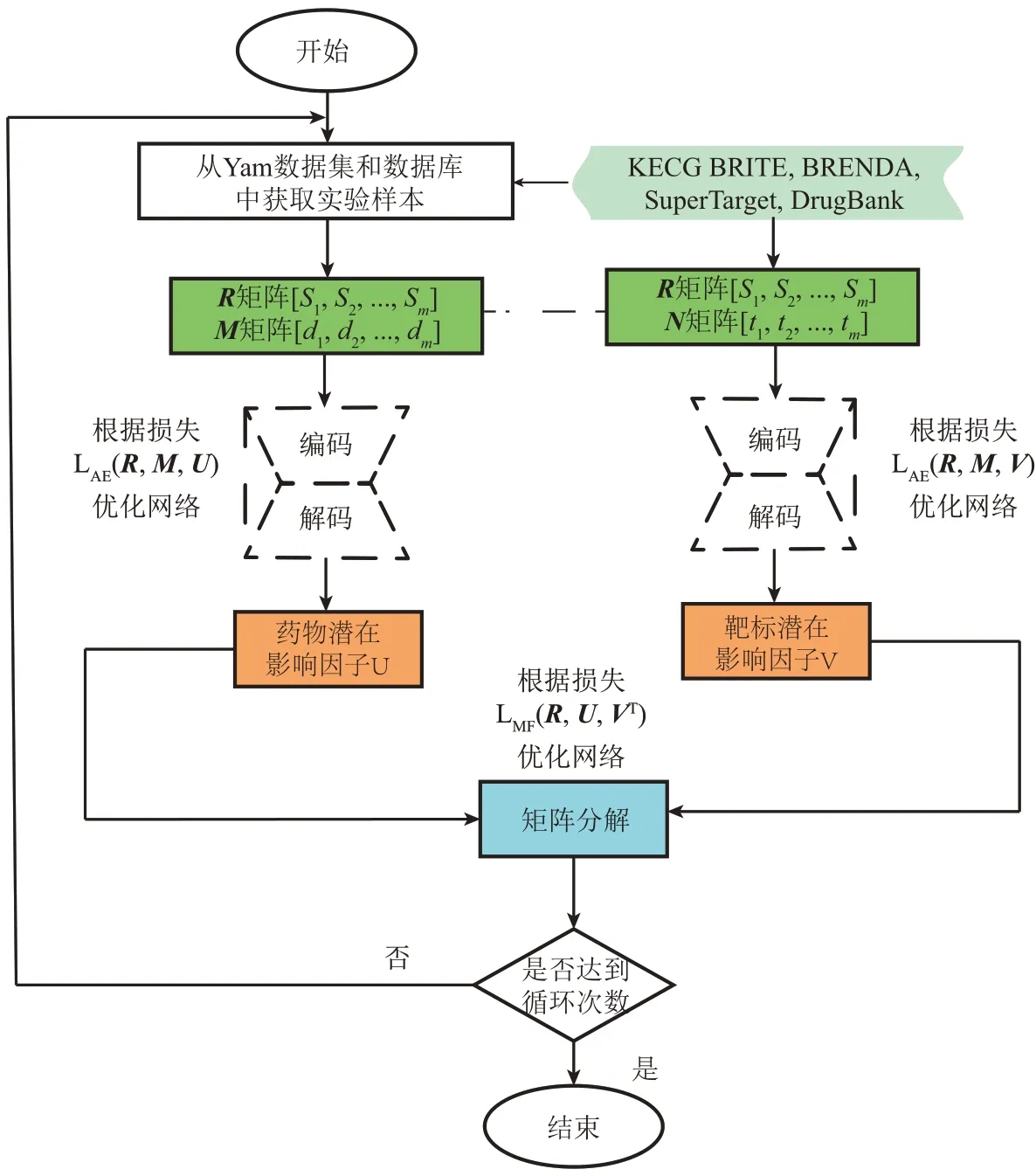
Fig.4 Deep collaborative filtering flowchart图4 深度协同过滤模型流程图
3.3 传统机器学习应用于药物活性预测小结
传统机器学习算法相较于传统基于实验的方法效率更高、耗时更短。DT、RF、SVM和矩阵分解等常用的机器学习模型对样本规模不敏感,只需要几千个训练数据样本即可训练出快速收敛的模型。药物研发领域往往由于客观原因导致实验数据较少,在此情形下传统机器学习模型依然可以实现有效建模。分析现有科研文献发现RF、半监督SVM和协同过滤在多数实验场景下拥有相对较高的药物靶标预测精度。但由于模型的适用范围有限、不同的模型适用不同的数据集,所以脱离实验数据和应用场景的模型好坏评价不能一概而论。
传统的机器学习模型在药物活性预测上效果较好,但是随着数据量的增加,机器学习模型的性能开始下降,也难以学习出复杂的函数模型。同时,传统机器学习类模型往往无法直接把实验数据属性变量输入模型,需要预先进行特征提取,对于高维的输入变量需要进行PCA等降维处理,才能进一步作为模型的输入。这些要求科研工作者需要有较好的数理统计基础及对常用数据预处理工具的熟练使用能力。
4 深度学习应用于药物活性预测
深度学习是机器学习中的一种,是当前机器学习领域最热门的研究方向。深度学习多被用于解决传统机器学习算法不擅长的具有高维输入变量的问题或复杂分类问题。对于类似于药物活性预测这样的复杂问题,深度学习通过学习数据样本的特点,自动寻找药物分子的特征而无需手动设计;也可以通过增加神经网络中的隐藏层进而提取更深层次的特征,显著提升模型的预测能力和分类能力。
目前深度学习在推荐、分类、目标检测、语音识别、图像分割、药物活性预测等方面取得了较大的突破。常见的深度学习模型有:多层感知机(multilayer perceptron,MLP)[65]、DNN[66]、卷积神经网络(convolutional neural networks,CNN)[67]、AE、循环神经网络(recurrent neural networks,RNN)[68]、长短期记忆神经网络(long short-term memory,LSTM)[69]、深度置信网络(deep belief networks,DBN)[70]、GAN[71]等。
4.1 常用深度学习模型简介
近年来,深度学习类模型层出不穷,但是大部分模型是在经典模型上的改进或者多模型间进行融合。本部分内容主要对被应用于药物活性领域的深度学习算法原理进行简要介绍,作为后续方法应用部分的理论铺垫和术语诠释。按照深度学习领域大的分类依序介绍,包含有ANN、DNN、AE、CNN、RNN、DBN和GAN等主要类型的深度学习算法。
ANN模型于1943年由神经生理学家McCulloch和数学家Pitts提出[72]。ANN中比较典型的是BP(back propagation)神经网络[73]。BP神经网络主要包含输入层(input layer)、隐藏层(hidden layer)和输出层(output layer)3个部分。其中,输入层接收外部信息并将信息输入模型,隐藏层对输入模型的信息进行处理,输出层负责信息的进一步优化和输出。BP神经网络的拓扑结构如图5所示。
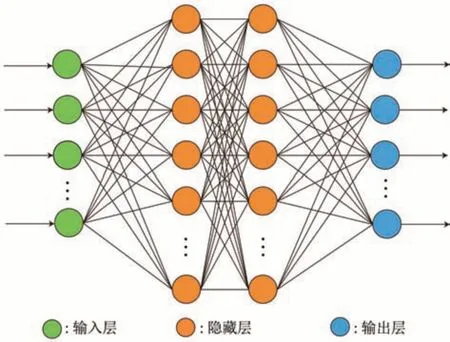
Fig.5 Neural network topology图5 神经网络拓扑结构
图中数据信息从绿色节点输入,最终从蓝节点输出,橙色节点为隐藏层节点,隐藏层的数量大于等于1且隐藏层之间依靠神经元的联结权重进行连接。BP网络的反向传播采用梯度下降算法对网络的整体结构进行修正,使得模型的损失函数达到最小。当神经元接收到的信号超出阈值时,神经元会被激活,然后通过激活函数的处理产生神经元的输出。常见的激活函数有阶跃函数、Tanh函数、Sigmoid函数和ReLU函数。其中阶跃函数由于具有不连续和不光滑的性质,故不常使用。
DNN概念宽泛,广义上CNN、RNN、GAN等都属于DNN。但通常提到DNN往往是指基于ANN和MLP进行拓展的拥有多隐藏层的全连接神经网络。其神经网络拓扑结构中隐藏层可高达几十上百层。
CNN是包含卷积计算的前馈神经网络,模型同时具有表征学习和稀疏表示的能力,在计算机视觉应用领域表现突出。具有代表性的CNN模型为Lecun等[67]于1998年提出的LeNet-5网络模型。该模型包含了现代CNN网络所具备的基本模块:卷积层、非线性激活层、池化层以及全连接层。在CNN模型中,卷积层内每个神经元都与其前一层中位置接近区域的多个神经元相连,该区域称为感受野(receptive field),其大小取决于卷积核的大小。卷积核根据设定的步长对输入的特征图进行扫描,在感受野范围内对输入特征矩阵做矩阵元素相乘求和并叠加偏移量。其原理见图6,在示例中输入的特征向量大小为5×5,卷积核的大小为3×3,步长为1;卷积核在输入向量构成的矩阵上移动,进行卷积运算实现降采样。近几年,新的CNN模型不断涌现,比较常见的CNN模型有:AlexNet[74]、VGGNet[75]、U-Net[76]、RBCNN[77]、YOLO[78]等。
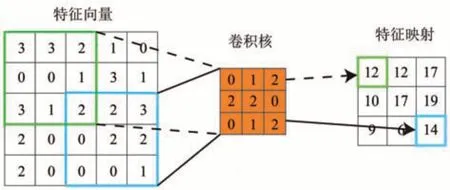
Fig.6 Diagram of convolution operation图6 卷积运算示意图
AE是一种无监督的神经网络,网络中的每个隐藏层都有一个编码器和一个解码器。编码器将输入变量编码为低维空间的特征变量;解码器对特征变量重新解码成高维特征变量。采用距离函数度量输入和输出信号之间的偏差损失。在解码的过程需要保证距离函数最小。
RNN又称递归神经网络。该模型适用于处理序列数据,其特点是在序列的演进方向进行递归且所有节点按照链式连接。故该模型多用于处理语音识别、机器翻译、价格预测、气象预测等问题。RNN原理如图7所示,在时刻t,输入信息为Xt,结合上一隐藏层的状态St-1和该隐藏层的状态St,输出信息Ot,矩阵W为隐藏层之间的权重,矩阵U为输入层与隐藏层之间的权重,矩阵V为隐藏层和输出层之间的权重。常见的RNN模型有LSTM[69]、GRU[79]、BRNN[80]等。
DBN是一个概率生成模型,与传统的判别模型神经网络相比,生成模型建立观察数据和标签之间的联合分布。其用于监督学习可使样本分类尽可能的准确,用于非监督学习可在保留原始特征同时降低特征维度。DBN模型架构由一个可视层和多个RBM层组合而成,层与层之间的神经元存在连接但层内的单元间不连接。其网络模型如图8所示。
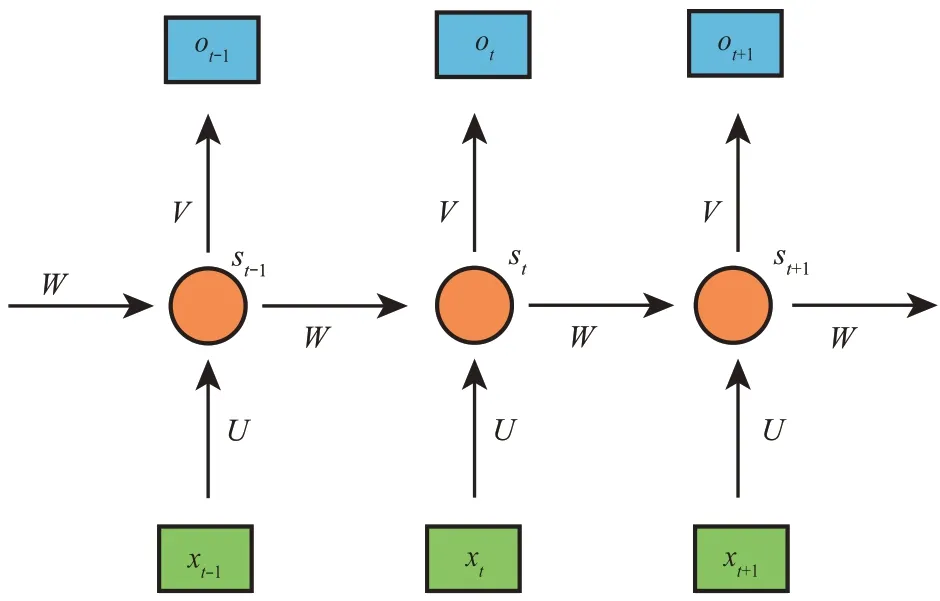
Fig.7 Diagram of principle of RNN图7 RNN原理示意图
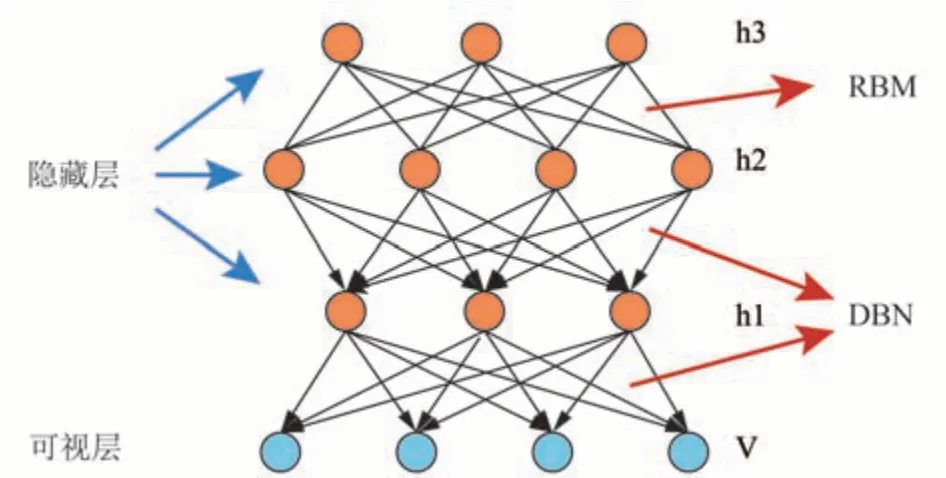
Fig.8 DBN network model图8 DBN网络模型
GAN是由Goodfellow等[71]提出的能够根据输入数据概率分布生成新数据的网络模型。GAN的隐藏层由两部分组成,分别是生成器和判别器。生成器用来产生新的样本,也称作假数据,判别器用来区分真实数据和假数据。模型经过多次迭代后,生成器生成数据的质量不断提升,直到判别器无法区分真实数据和假数据。
使用GAN模型能够产生大量可供训练的数据样本,但是在模型训练过程中易出现梯度消失问题。此外,当生成器生成质量差的样本且通过增加训练时间也无法改善质量时,GAN会出现模式崩溃[81]。条件生成对抗网络(conditional generative adversarial networks,CGAN)[82]可以对GAN模型进行调节避免模式崩溃。另一个改进的模型Wasserstein GAN(WGAN)[83]通过引入EM距离,并缩小EM距离对生成器和鉴别器进行优化,使得生成数据概率分布接近真实数据概率分布。Wang等[84]把EM和GAN相结合提出了进化生成对抗网络(evolutionary generative adversarial networks,E-GAN),解决了GAN模型训练不稳定及模式崩溃的问题。
4.2 深度神经网络在药物活性预测中的应用
深度神经网络类算法一般不需要手动提取数据特征。在药物活性预测领域,深度网络能处理较为复杂的药物分子数据,通过逐层抽象及传递,模型可以自动提取较深层次的数据特征,进而获得更高的药物活性预测准确率。本部分内容对当前深度神经网络在药物活性预测领域中的应用研究进行分析总结,主要涉及DNN、CNN、RNN、DBN、GAN等深度学习模型的应用(表3)。
4.2.1DNN在药物活性预测中的应用
DNN是深层全连接神经网络,在很多应用场景下效果良好。Bharath等[85]借助DeepChem开源平台构建了多任务DNN、单任务DNN、Progressive DNN、Bypass DNN和RF,共5类模型。实验使用默沙东公司2012年在Kaggle平台上举办的药物活性预测比赛的4个数据集Kaggles、Factors、Kinase、UV进行训练和测试。在4个数据集上的实验结果表明,多任务DNN模型在训练集上的R2可能会低于RF以及另外4种DNN模型,但在验证集和测试集上能够取得比另外4种模型更高的预测精度。此外其他3种DNN模型的预测效果均优于RF。在Kaggle数据集上,多任务DNN在训练集和测试集上能达到0.793和0.468的精度,而RF能达到0.941和0.428。由此可知在药物预测方面DNN模型相较于以RF为代表的机器学习有着较好的泛化能力和更高的预测精度,同时DNN中多任务DNN的预测精度和泛化能力又高于其他类型DNN模型。
同Bharath等的工作类似,Cai等[86]提出了DeephERG框架来构建hERG通道阻滞剂评估的预测模型。内置DeephERG的多任务DNN算法显示出令人满意的预测结果,在验证集的AUC值为0.967,优于单任务DNN的0.957,RF的0.950,SVM的0.908,朴素贝叶斯的0.922以及图卷积神经 网 络 (graph convolutional neural network,GCNN)的0.959。在单任务DNN应用方面,Kato等[87]通过对单任务全连接神经网络(fullyconnected deep neural network,FC-DNN)进行调超参,搭建了新的QSAR/DNN模型,取得了同Kaggle QSAR竞赛冠军组模型相近的R2值。路莹莹[88]在DrugBank数据集上采用FC-DNN进行药物靶标相互作用预测。实验结果表明,FC-DNN的AUC和准确率分别为0.96和0.88,对照组模型RF和SVM对应的AUC及准确率分别为0.90、0.84和0.92、0.85。FC-DNN模型在测试集上的准确率和AUC都优于对照组的传统机器学习模型。
4.2.2CNN在药物活性预测中的应用
CNN模型常用于处理基于图像识别的问题或类图像识别的问题。在药物活性预测研究领域,药物分子结构可以用二维或三维的图像来表示,CNN通过对分子图像识别的方式来识别分子特征及类型。Chemception[89]和AugChemception[90]模型分别使用化合物的分子结构图像进行化学性质预测,两种方法都采用了类似Inception-ResNet-v2网络的构造。Chemception模型通过对化合物的2D分子图像进行学习,能够对化合物的化学性质进行预测,预测流程如图9所示。该模型使用机器学习基准数据集MoleculeNet中的3个基准数据集Tox21、HIV、FreeSolv进行验证,采用分子结构绘制软件RDKit将SMILES格式的结构数据绘制成80×80像素的二维分子图像作为输入,与输入传统分子特征相比,Chemception模型在HIV训练集和测试集上AUC分别为0.796、0.798,RMSE分别为1.17、1.22 kcal/mol,优于以分子特征作为输入的多层感知器深度网络模型。
在药物靶标活性预测中,胡姗姗[91]在LeNet-5网络的基础上进行改进,将药物小分子和靶标蛋白的近邻特征映射成为矩阵数据。其改进后的模型在来自KEGG BBRITE数据库的数据集上AUC值为0.952 7,准确率为0.881 4,比DBN分别高3.69%和2.26%。Kun等[92]使用CNN模型实现了基于3D小分子的电子密度数据进行碳氢化合物的Kohn-Sham动能和KS密度预测,进一步对传统动力学函数进行局部校正。实验结果表明,CNN能够半定量的预测键合并准确预测KS分子动力学轨迹。另外含3层卷积层的CNN模型F误差总体低于含有2层或者4层的模型。在烷烃分子动能数据集上,模型在训练集和测试集上结果误差可分别达到约0.01和0.08。同时模型能够克服数据中的非线性噪声,作者认为可以通过加强物理约束、提升数据精度来提升模型的学习能力。
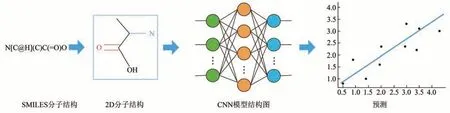
Fig.9 CNN drug activity prediction flowchart图9 CNN药物活性预测流程图
4.2.3AE在药物活性预测中的应用
AE将输入的高维数据映射到潜在空间并用较低维度表示,处理后的数据被重新解码成原始高维输入形式。其降维功能便于处理大量高维数据,同时提高模型准确性。
Rafael等[93]采用变分自编码器(variational autoencoder,VAE)来生成化合物的结构。编码器将离散的分子SMILES字符串被映射到潜在空间中,并通过隐向量进行连续表示。用z来表示分子结构的连续表示,则模型中的多层感知模块训练出函数f(z)来预测新的分子结构,并采用梯度优化方式对f(z)进行优化进而寻找新的满足医学特征需求的分子结构。该模块生成新的化学结构的方式包括解码随机向量、扰乱已知的化学结构和在分子之间进行插值。解码器将编码器输出的连续表示逆向转化为离散的SMILES字符串。在不同的数据集上神经网络采用不同的超参数:
在ZINC数据集上的模型超参数配置如下。a.编码器:三层一维卷积层(过滤器大小分别为9、9、10,数量分别为9、9、11);b.全连接神经网络:宽度为196;c.解码器:三层门控循环单元(GRU)网络,隐藏层维度为488。
在QM9数据集上的模型超参数配置:a.编码器:三层一维卷积层(每层滤波器大小分别为2、2、1,数量分别为5、5、4);b.全连接神经网络:宽度为156;c.解码器:三层门控循环单元(GRU)网络,隐藏层维度为500。
实验结果表明,Rafael等[93]构造的模型相较于其他常见机器学习模型具有更高的药物相似性定性估计值。随后,作者将高斯过程、高斯搜索和遗传算法引入预测模块对模型进一步优化。结果发现,相较于高斯搜索和遗传算法,高斯过程的引入对模型的预测效果提升具有明显的作用。
类似的工作,Thomas等[94]采用VAE模型生成了对2型多巴胺受体具有活性的新型化合物。闫奕霖[95]分别建立了基于SVM、ANN和SAE的化合物抗HIV活性分类预测模型并进行对比实验。在不同数据集上的结果均表明SAE能够实现化合物抗HIV活性分类更高的预测精度。
4.2.4RNN在药物活性预测中的应用
RNN将序列数据作为输入,在序列的演进方向进行递归且所有节点按照链式连接,故其多用于对序列概率分布进行建模。
Segler等[96]采用具有3个堆叠LSTM层的RNN模型进行药物结构设计,并经过迁移学习生成对所需生物靶标具有良好亲和力的新型分子。模型设计流程如下:a.采用目标预测模型(RF、逻辑回归、梯度提升树、DNN)对药物分子活性进行预测;b.迁移学习,对预训练的RNN模型进行小样本重新训练;c.采用目标预测模型对上一步骤设计药物进行活性预测,然后将预测为具有一定活性的药物合并入步骤a,形成闭环。
模型采用随机富集(enrichment over random,EOR)方法进行评估,EOR表示如下:
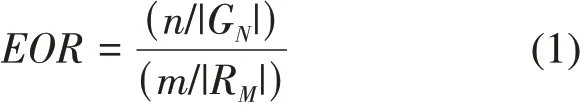
公式中,n=|G N∩T|是测试集T和微调模型生成的药物分子集合G N的并集元素的数量,m=|R M∩T|,RM是在大数据集合上训练的通用模型生成的药物分子集合。在金黄色葡萄球菌药物数据集上,迁移学习训练的模型跟从头训练的模型相比,预测结果误差更小。同时上述循环模型迭代8次之后EOR值为59.6,高于仅进行单次简单重训练模型(EOR=6.3)。
Bjerrum等[97]基于带有LSTM结构的RNN模型建立序列到序列异源编码器对SMILES预测不同的枚举字符串。实验表明,异源编码器派生的向量明显优于自编码器和使用ECFP4指纹建立模型的派生向量。龙飞达[98]基于PubCHEM数据库采用具有3层门控结构的RNN结合马尔可夫决策过程(Markov decision process,MDP)[99]在靶标蛋白CRM1抑制剂莱菔素的基础上进行分子结构改造,将改造后得到的CRM1靶向小分子抑制剂命名为LFS-1107。活性测试实验表明该分子对CRM1具有更高的药物活性。路莹莹[88]采用LSTM在DrugBank数据集上进行药物靶标相互作用预测实验,以配体结构信息和蛋白质序列信息构建药物靶标相互作用向量作为模型输入,结果表明LSTM模型的预测准确率和AUC分别为0.87和0.95,皆高于传统机器学习算法RF和SVM。
4.2.5DBN在药物活性预测中的应用
在DBN预测药物活性的应用中,首先将药物特征输入其模型前端RBM网络,然后将RBM网络的输出作为BP神经网络的输入,进一步对特征进行深层提取,最后输出药物活性的预测类别。
高双印[61]从PubChem上选取3类药物活性数据,PLK1 PBD、SMAD3和IL-1B,并对上述3类蛋白质结构数据进行SVM、ANN、SAE和DBN建模实验。测试结果表明,在3种数据集上,无监督算法SAE和DBN在准确率、召回率、马修斯相关系数等评价指标上均优于另外两种算法。其中,在PLK1 PBD数据集上(仅含2D分子描述符),DBN表现最好,在输入特征数为50,隐藏层数为3时模型精度达到最高(90.1%);在IL1-B数据集上,训练集为3DI、3DX和ALL分子描述符时,DBN预测表现优于SAE和其他模型,训练集为2D分子描述符的时候,DBN预测表现略逊色于SAE;在SMAD3数据集上,模型输入特征数为125,隐藏层为1层,在准确率、召回率、误分率等评价指标上,SAE均优于DBN,其中SAE的准确率达到93.38%,DBN达到83.81%。由此可见模型预测效果好坏不仅与模型本身有关,也与数据集有关。
同样的,黎佳朗[15]比较了DBN、SAE结合SVM、基于网络的推荐算法和矩阵分解方法对药物靶标相互作用进行预测,实验结果表明DBN相较于其他模型具有更高的准确率。
4.2.6GAN在药物活性预测中的应用
GAN中的生成器和判别器不断进行博弈和多次迭代逐步提升生成器生成数据的质量,直至判别器无法区分真实数据和假数据,从而可以生成大量可靠的训练样本。
Cao等[100]提出了MolGAN模型,一个隐式的无似然的小分子图生成模型,它避开了基于似然的复杂计算图匹配过程和节点排序启发式的需要。该模型的工作流程如下:a.生成器根据先验分布生成邻接张量A和注释矩阵X;b.对A和X进行采样稀疏化,然后合并成注释分子图;c.基于图卷积神经网络(GCN)的鉴别器和奖励网络对分子图进行处理,同时不会改变图中节点的排列顺序。
实验结果表明,强化学习的加入使得生成的分子结构有效性由87.7%提升至99.8%,分子的新颖性和溶解度也随着强化学习模块所占比重的增加而提高。在QM9数据集上,MolGAN相较于不同的VAE模型和ORGAN(objective-reinforced GAN)取得了较好的预测结果,同时运算速度比ORGAN模型快近5倍。MolGAN主要优势为将GAN和强化学习结合,同时直接针对分子图进行训练而不是生成的序列化数据。但是该模型存在模式崩溃的可能,同时生成的分子多样性相较于另外两类模型较差。提出者认为将来可以通过改进奖励函数的设计或者引入其他的预训练模型来避免此类问题。
陈浩宇[101]将GAN模型结合其他生成器目标并进行优化,同时引入进化算法思想,对生成器的目标函数进行改造,获得稳定且生成化合物质量更高的模型MDEGAN。最终获得的生成器目标为:
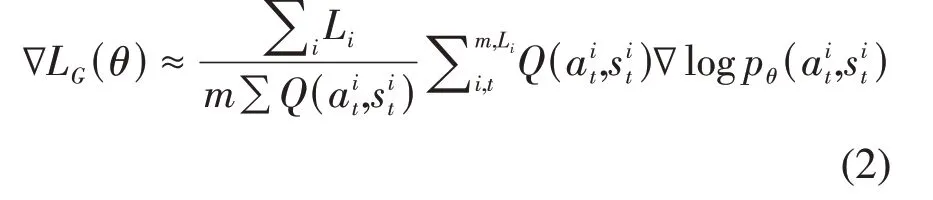
其中Q(a,s)表示给定前代生成s进行的生成动作a。进化算法的适应度函数为:
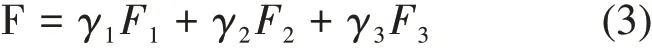
式中F1、F2、F3分别表示溶解性、可合成性以及类药性的环境适应度函数,γ≥0,用来平衡3个指标。生成器目标优化通过加强约束提高药物生成质量,而进化算法则增加药物多样。实验结果表明,同其他传统的GAN模型相比较,MDEGAN可以生成更具多样性和药物特性且质量更高的化合物序列。
4.2.7其他改进深度学习模型在药物活性预测中的应用
耦合强化学习、GNNs、注意力机制等的深度学习模型近几年发展较快,也被应用于药物活性预测。
Zheng等[102]通过LSTM模型提取了基因数据和药物数据之间潜在的语义信息;然后构造CNN模型,以LSTM的输出作为CNN模型的输入,实现基因组数据的提取;最后使用全连接网络进行预测,该模型取得了较好的性能。Lee等[103]提出了一种基于CNN和DNN的模型——DeepConv-DTI,在原始蛋白质序列上使用CNN以捕获广义蛋白质类别的局部残基模式,从而取得更好的预测结果。Guimaraes等[104]将GAN与强化学习结合,并引入奖励机制。该模型在生成编码为文本序列(SMILES)分子的同时能够有效地将生成过程偏向期望的指标。
Xiong等[12]为药物分子表示引入了一种新的GNNs架构Attentive FP,该方法使用图注意力机制从开源的药物数据集中学习分子内部的相互作用。相较于基于图模型的分子表示方法,Attention FP模块在评估邻接原子的影响时更加有效。结构上,Attention FP模型内部包括目标原子在某一时刻的状态向量和注意内容向量,联结节点的权重越高,对应分子图上的虚线颜色越深。其对应的联结、权重、语境输出公式如下所示:
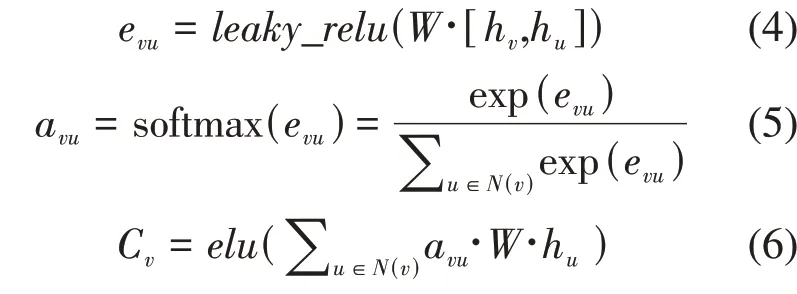
其中,[h v,h u]是目标节点和邻居节点的联合状态向量,C v为节点的文本向量,W为网络训练得到的权重。
在PDBbind和QM9数据集上,Attentive FP在溶解度、疟疾生物活性和光伏效率上取得了比Neural FP、Weave和MPNN更好的预测效果。赵其昌[105]提出一种端到端的基于图注意卷积和交叉注意力机制的药物靶标相互作用预测模型,以及一种端到端的基于CNN和协同注意力机制的药物蛋白质亲和力值回归模型。以上两种模型均是通过CNN模块提取特征矩阵,使用注意力机制强化语义信息在模型中的重要程度。路莹莹[88]在DrugBank数据集上采用高速路神经网络(highway network,HN)对药物靶标相互作用进行预测。通过增加门限机制,将输入网络模型的部分通路数据进行非线性转换,该机制能够有效缓解梯度爆炸和梯度消失问题。模型准确率和AUC分别达到0.88和0.94。
4.3 深度学习应用于药物活性预测小结
近年来,深度学习模型被逐步应用于药物活性预测中。其中DNN模型应用最为广泛,成熟度高,效果也比较好,拥有广阔的应用前景;CNN由于其具有卷积和池化功能,常用于解决类似于图像识别及分类的问题,例如基于药物分子结构的二维图像进行分子类别判断;AE具有较好的高维数据处理能力,可有效解决繁杂药物分子和靶标蛋白的特征表示;RNN模型具有连接的循环单元,故适用于处理如自然语言处理、蛋白质氨基酸序列推断等具有时间序列类数据的问题,常应用于氨基酸序列的预测和推断,常用的RNN模型为LSTM和BRNN;DBN采用二进制变量对数据概率分布进行估计,进而建立数据和标签之间的联合分布,该类模型在药物活性预测中应用较少,但是在部分3D分子描述符数据集上训练效果优于AE;GAN模型能够生成新的数据,在训练数据样本较少的情况下可以通过增加样本数量来提高模型的预测精度。
深度学习模型中较为常用且效果较好的模型为DNN、CNN、RNN和AE。很多学者通过对不同模型进行结合使用,取得了较好的实验结果。目前更多的研究人员倾向于将迁移学习、强化学习、注意力机制等引入深度学习模型,以此来缩短训练时间、提高模型精度,避免无关因素的干扰。大量的实验结果表明,迁移学习、注意力机制等技术的引入很大程度上提高了药物分子的预测精准度以及药物分子生成的有效性。
5 总 结
随着生物、医学技术的不断发展,药物分子活性数据以及医学临床数据不断扩增,药物研发的重点逐渐向海量生化大数据挖掘的方向发展。以基于配体的预测方法为例,近几年结合传统机器学习、矩阵分解和深度学习等的方法逐渐受到业界的广泛关注并被寄予厚望。随着生物医药生产及临产相关各类数据的多样化和计算机计算能力的逐步增强,生物活性预测方法逐步引入深度学习模型、GNNs模型等最新算法模型,其在药物研发领域发展迅速并大幅缩短了药物研发时间,降低了药物研发成本。
药物活性预测和药物设计由纯生化实验到机器学习算法助力再到深度学习算法应用的过程中,药物分子活性预测的效率不断提升。由上述内容可以得知,与有监督或者半监督的机器学习方法(SVM、RF等)相比,无监督的深度学习(FCDNN、CNN、RNN、SAE等)能够更加全面的对药物分子结构进行学习并且给出更加准确的预测结果。但深度学习模型有时会出现过拟合现象,即在训练集预测精度很高,在测试集上预测精度比较低,或者在部分数据集上预测精度较高,在其他数据集上预测精度较低,亦或是出现网络梯度爆炸或梯度消失的问题,导致模型无法对数据样本进行有效学习和进一步应用。数据预处理、算法创新及模型结构的改进优化将会成为未来计算机辅助药物活性预测的主要任务。
6 展 望
目前,深度学习模型在药物分子活性预测及相关方向取得了较好的进展,其中DNN、AE以及RNN模型在该领域应用的较为广泛。有研究者也尝试利用其他最新的算法模型应用于药物活性预测。未来有望开展的研究有如下几个方面:
a.强化学习与深度学习结合应用于药物活性预测。强化学习的原理是对人或动物学习过程的模仿,通过设计有行为、感知、奖励的循环流程来强化正确行为,惩罚错误行为。强化学习与传统有监督机器学习模型的拟合方法有很大不同。模型训练过程中每一个新反馈都被立即传递,完成一次参数迭代,故而拥有较快的学习速度。强化学习相较于传统模型提供了更快的学习机制,并且鲁棒性较强。当前,在图像目标检测和自然语言处理领域,结合强化学习的深度学习模型取得了优异的成果。进一步探索其在药物活性预测领域的应用,或许有意想不到的收获。
b.多模型联用集成应用于药物活性预测。集成学习是机器学习中一类学习算法,其主要通过对多个学习器进行训练和组合实现比单个学习器更好的预测结果。传统的机器学习模型以及各类深度学习模型在解决某类问题时有各自的优势,对于一个复杂的问题来说,单独使用其中一种模型无法达到理想的预期结果。但是集合多个不同类的预测模型进行集成学习往往会取得更好的结果。在现有的探索基础上,借助模型集成思路解决药物活性预测领域的相关难题也是一个可选的研究方向。
c.从生物医药工程的角度提出新的模型或模式。在解决或助力于解决行业问题时,算法或技术只是充当工具的作用,算法工具是否可以发挥大的威力很大程度上决定于使用者对具体行业的专业认知,以及进一步对问题的准确建模。所以,药物活性预测的发展依然离不开医药行业的专业认知。故应该从生物医药工程角度对药物设计的各个环节不断地提出新的问题,进一步构建有效的模型来解决问题,推动药物研发的自动化和智能化。
d.深度学习预训练大模型应用于药物活性预测。2017年谷歌Transformer深度网络结构的提出,使得深度学习模型参数突破了1亿,BERT网络模型的提出,使得深度学习模型参数量进一步超过3亿规模。2020年,OpenAI公司的GPT-3模型参数达到1 750亿,随后谷歌Switch Transformer的问世使得模型参数一举突破万亿规模。截止2021年底,北京智源的悟道、阿里达摩院的M6和快手的CTR等大模型参数量均突破万亿。大模型的应用有助于推动多个领域的难题突破。例如,谷歌DeepMind团队相继推出的AlphaFold和AlphaFold2模型被应用于蛋白质结构预测,后者的蛋白质结构预测准确性接近了真实结构。大模型的应用有望进一步在药物活性预测领域实现重大进展。
