端烷氧基聚甲基三氟丙基硅氧烷的合成表征和室温交联特性
2022-08-01刘润竹张天福张孝阿王成忠江盛玲
刘润竹,张 猛,张天福,张孝阿,王成忠,江盛玲,史 翎,李 伟
(1. 北京化工大学碳纤维及功能高分子教育部重点实验室材料科学与工程学院,北京 100029;2. 航天化学动力技术重点实验室湖北航天化学技术研究所,湖北襄阳 441003)
氟硅密封胶的基础聚合物为低分子量氟硅均聚物或共聚物,其主链为Si—O 结构,侧基含三氟丙基,具有优异的耐高低温(-60~200 ℃)、耐非极性化学介质和耐老化性能,在飞机整体油箱密封等领域具有重要应用价值[1]。氟硅均聚物的三氟丙基单元的摩尔分数为100%,具有最为优异的耐燃油性能,以此为基础制备的单组份脱酸型氟硅密封胶已在工业部门获得应用,然而中性缩合体系的交联反应速率较差,影响了进一步的应用[2]。
为了进一步改善氟硅密封胶的耐高温性能和交联性能,研究者在基础聚合物改性、交联剂和耐热添加剂优选等方面作了许多工作。将亚苯基引入氟硅聚合物主链中,能够显著改善所得聚合物的耐高温性能,然而其玻璃化转变温度有所提高,对低温性能不利[3]。采用双子型硅氧烷交联剂,如1,4-双[三甲氧基硅烷基(乙基)]苯(BTMSEB)、1,2-双(三乙氧基硅烷基)乙烷(BTSE)和1,6-双(三甲氧基硅烷基)己烷(BTMSHEX),得到的硫化胶具有比传统脱醇型交联剂正硅酸乙酯(TEOS)更好的耐热性[4]。鉴于大体积三氟丙基侧基对相邻的端羟基有很大的屏蔽作用,会对交联反应性和硫化性能造成不利影响。利用二甲基硅氧烷链段在氟硅均聚物两侧进行封端,能够得到ABA 三嵌段聚合物,虽然解决了氟硅均聚物硫化速率慢的问题,但降低了三氟丙基单元含量,不利于耐油性[5]。
为了兼顾氟硅低聚物的端基反应性和三氟丙基含量,通过小分子含氢硅氧烷对氟硅均聚物进行封端,既能隔开三氟丙基侧基与反应性端基的距离,减小屏蔽作用而改善交联反应性,又不会显著降低三氟丙基单元含量。基于此,本文以α,ω-端羟基聚甲基三氟丙基硅氧烷为初始聚合物,采用多种含氢硅氧烷对其进行封端(如Scheme 1),表征了封端聚合物的结构。通过流变学方法探究了封端聚合物交联硫化过程中的黏度(η)和模量(G',G'')的变化趋势,研究了聚合物相对分子质量、烷氧基类型和数量等因素对交联反应速率的影响。
1 实验部分
1.1 原料与试剂
α,ω-端羟基聚甲基三氟丙基硅氧烷(H1~H4):黏度分别为145 mPa·s,5000 mPa·s,11200 mPa·s 和21000 mPa·s,威海新元化工有限公司;三甲氧基硅烷(TMS)和甲基二甲氧基硅烷(MDMS):98%,阿拉丁试剂公司;钯碳催化剂:钯的质量分数5%,含水40%~60%,阿拉丁试剂公司;气相二氧化硅(R8200):赢创德固赛公司;氧化铁红:工业级,上海一品公司;正硅酸乙酯(TEOS,98%)、二月桂酸二丁基锡(97.5%)、四氢呋喃和三乙胺、三乙氧基硅烷(TES,97%):北京百灵威科技有限公司。
1.2 端烷氧基聚甲基三氟丙基硅氧烷的合成
α,ω-端羟基聚甲基三氟丙基硅氧烷与不同的含硅氢硅氧烷进行封端反应,得到多种端烷氧基聚甲基三氟丙基硅氧烷(Scheme 1),包括三甲氧基封端(M1,M2,M3,M4)、甲基二甲氧基(M5)和三乙氧基封端(E1)3 类聚合物。

Scheme 1 Synthesis of alkoxy-terminated poly(methyltrifluoropropyl)siloxanes
以M1 的合成为例。500 mL 三口烧瓶配上回流冷凝器和机械搅拌器,通入氮气,在烧瓶中将5.9 g(0.01 mol)的α,ω-端羟基聚甲基三氟丙基硅氧烷(H1)溶解于350 mL 的四氢呋喃中,再加入3.67 g(0.03 mol)TMS 和0.4 g 的钯碳催化剂,室温搅拌混合均匀。缓慢加热至60 ℃,观察到因氢气生成而产生了大量气泡,继续加热反应8 h,结束反应。过滤除掉催化剂,减压除去溶剂和其它可挥发性物质,得到淡黄色透明黏稠液体状目标产物(M1),产率92%。Mn= 930,Mw/Mn=1.59 (GPC, Polystyrene calibration)。 FT-IR,ν(cm-1): 2951, 2911, 2849, 1466,1422, 1369, 1316, 1267, 1211, 1126, 1091, 1071, 1028,901, 838, 767, 637, 552;1H -NMR (400 MHz, acetone-D6),δ(TMS): 3.55~3.62 (—OCH3), 2.22~2.30 (—CH2—CF3), 0.85~0.92 (Si—CH2—), 0.25~0.32 (—Si—CH3);13C- NMR (100 MHz, acetone-D6),δ(TMS):123.84~132.06 (—CF3), 50.21~50.28 (—OCH3), 26.82~27.79 (—CH2—), 8.21~8.74 (Si—CH2—), (-2.35) ~(-1.79) (Si—CH3)。
采用类似反应步骤也可合成了M2,M3,M4,M5 和E1,物料种类和加入量如Tab. 1 所示。

Tab. 1 Amount of reactants added for synthesizing of M2,M3,M4,M5 and E1
M2 的表征数据:淡黄色透明黏稠液体,产率94% 。Mn=9400,Mw/Mn=1.82 (GPC, Polystyrene calibration)。 FT-IR,v(cm-1): 2951, 2911, 2849, 1466,1422, 1369, 1316, 1267, 1211, 1126, 1091, 1071, 1028,901, 838, 767, 637, 552;1H-NMR (400 MHz, acetone-D6),δ(TMS): 3.55~3.62 (—OCH3), 2.22~2.30 (—CH2—CF3), 0.85~0.92 (Si—CH2—), 0.25~0.32 (—Si—CH3);13C- NMR (100 MHz, acetone-D6),δ(TMS):123.84~132.06 (—CF3), 50.21~50.28 (—OCH3), 26.82~27.79 (—CH2—), 8.21~8.74 (Si—CH2—), (-2.35) ~(-1.79) (Si—CH3)。
M3 的表征数据:淡黄色透明黏稠液体,产率95% 。Mn=13500,Mw/Mn=2.48 (GPC, Polystyrene calibration)。 FT-IR,v(cm-1): 2951, 2911, 2849, 1466,1422, 1369, 1316, 1267, 1211, 1126, 1091, 1071, 1028,901, 838, 767, 637, 552;1H-NMR (400 MHz, acetone-D6),δ(TMS): 3.55~3.62 (—OCH3), 2.22~2.30 (—CH2—CF3), 0.85~0.92 (Si—CH2—), 0.25~0.32 (—Si—CH3);13C- NMR (100 MHz, acetone-D6),δ(TMS):123.84~132.06 (—CF3), 50.21~50.28 (—OCH3), 26.82~27.79 (—CH2—), 8.21~8.74(Si—CH2—), (-2.35) ~(-1.79) (Si—CH3).。
M4 的表征数据:淡黄色透明黏稠液体,产率95% 。Mn=19200,Mw/Mn=2.22 (GPC, Polystyrene calibration)。 FT-IR,v(cm-1): 2951, 2911, 2849, 1466,1422, 1369, 1316, 1267, 1211, 1126, 1091, 1071, 1028,901, 838, 767, 637, 552;1H-NMR (400 MHz, acetone-D6),δ(TMS): 3.55~3.62 (—OCH3), 2.22~2.30 (—CH2—CF3), 0.85~0.92 (Si—CH2—), 0.25~0.32 (—Si—CH3);13C- NMR (100 MHz, acetone-D6),δ(TMS):123.84~132.06 (—CF3), 50.21~50.28 (—OCH3), 26.82~27.79 (—CH2—), 8.21~8.74 (Si—CH2—), (-2.35) ~(-1.79) (Si—CH3)。
M5 表征数据:淡黄色透明黏稠液体,产率93% 。Mn=12900,Mw/Mn=2.58 (GPC, Polystyrene calibration)。 FT-IR,v(cm-1): 2951, 2911, 2849, 1466,1422, 1369, 1316, 1267, 1211, 1126, 1091, 1071, 1028,901, 838, 767, 637, 552;1H-NMR (400 MHz, acetone-D6),δ(TMS): 3.50~3.54 (—OCH3), 2.19~2.32 (—CH2—CF3), 0.77~0.94 (Si—CH2—), 0.25~0.32 (End group —Si—CH3), 0.15~0.19 (Side group —Si—CH3);13C-NMR (100 MHz, acetone-D6),δ(TMS): 124.85~133.07 (—CF3), 50.07~50.16 (—OCH3), 27.54~28.94(—CH2—), 9.34~9.73 (Si—CH2—), (-1.05) ~(-1.77)(Side group Si—CH3), (-3.06) ~(-2.74) (End group —Si—CH3)。
E1 表征数据:淡黄色透明黏稠液体,产率94% 。Mn=13800,Mw/Mn=2.55 (GPC, Polystyrene calibration)。FT-IR,v(cm-1): 2978, 2931, 2899, 1445,1391, 1369, 1315, 1266, 1211, 1126, 1104, 1082, 1030,967, 900, 841, 796, 551;1H-NMR (400 MHz, acetone-D6),δ(TMS): 3.82~4.01 (—OCH2—), 2.18~2.34 (—CH2—CF3), 1.19~1.29 (—CH3), 0.85~0.91 (—Si—CH2—), 0.26~0.32 (—Si—CH3);13C-NMR (100 MHz,acetone-D6),δ(TMS): 125.11~133.32 (—CF3), 58.14~60.24 (—OCH2—), 28.22~29.13 (Side group—CH2—),18.67~19.25 (Si—CH2—), 9.82~10.09 (—CH3), (-1.01)~0.34 (Si—CH3)。
1.3 测试与表征
1.3.1 红外光谱(FT-IR)分析:采用Nicolet iS5 型FT-IR 傅里叶变换红外光谱仪(美国赛默飞公司)进行测试。KBr 压片溶液涂膜进行制样,扫描范围400~4000 cm-1。
1.3.2 核磁共振(NMR)分析:采用AV400 型核磁共振波谱仪(德国Bruker 公司)进行氢谱(1H-NMR)和碳谱(13C-NMR)测试。溶剂为氘代丙酮,内标为四甲基硅烷(TMS)。
1.3.3 凝胶渗透色谱(GPC)分析:采用515 型凝胶渗透色谱仪(美国Waters 公司)进行相对分子质量及其分布测试。测试温度30 ℃,THF 为流动相,流速1 mL/min,标样为聚苯乙烯。
1.3.4 流变性能测试:采用MCR-52 型流变仪(德国安东帕公司)进行黏度(η)和动态模量(G'和G'')测试。硫化体系配比:基础聚合物、交联剂、二月桂酸二丁基锡的质量比为100:4:1,样品盘温度和相对湿度设定为25 ℃和50%,转子与样品盘间距设为1 mm。其中黏度采用稳态旋转模式进行测试,模量采用动态振荡模式进行测试,对样品进行等温时间扫描,频率(ω)恒定为10 rad/s,应变(γ)恒定为1%。
1.3.5 交联密度测试:采用NMR C12-010V-T 低场核磁交联密度仪(纽迈公司)进行交联密度测试,通过测量样品的交联链、悬尾链和自由链的比例,进而计算出交联密度。
1.3.6 力学性能测试:采用CMT4204 型万能材料试验机(美特斯公司)进行拉伸性能测试,参照国标GB/T 528-2009,测试温度为(23±2)℃,试验机速率为200 mm/min。采用邵氏A 硬度计(扬州市源恒机械有限公司)进行硬度测试,参照国标GB/T 531.1-2008。
1.3.7 热失重(TGA)分析:采用SDT-Q600 型热失重分析仪(美国TA 公司)进行热稳定性试验,测试温区为室温至600 ℃,升温速率为20 ℃/min,气氛为氮气。
1.3.8 差示扫描量热(DSC)分析:采用DSC25 型差示扫描量热仪(美国TA 公司)进行玻璃化转变温度(Tg)测试,测试温区为-90 ℃至120℃,升温速率为10 ℃/min,氮气流速为50 mL/min。
2 结果与讨论
2.1 合成与表征
根据Scheme 1 中的合成路线,以4 种不同黏度的α,ω-端羟基聚甲基三氟丙基硅氧烷(H1,H2,H3和H4)为初始聚合物,选用3 种含氢硅氧烷(TMS,TES 和MDMS)为封端剂,在钯碳催化剂作用下进行脱氢缩合反应,制备了6 种烷氧基封端的聚甲基三氟丙基硅氧烷,分别为三甲氧基硅烷封端聚合物(M1,M2,M3 和M4)、二甲氧基硅烷封端聚合物(M5)和三乙氧基硅烷封端聚合物(E1)。Tab. 2 给出了不同封端聚合物的数均相对分子质量及多分散系数,可以看出,封端聚合物与它们的初始聚合物具有几乎相同的相对分子质量,这是封端反应的典型特征,结合脱氢缩合反应前后的GPC 淋出曲线(Fig.1),发现并无因扩链反应而产生的肩峰,排除了发生扩链反应的可能性,进一步证实了封端反应的成功
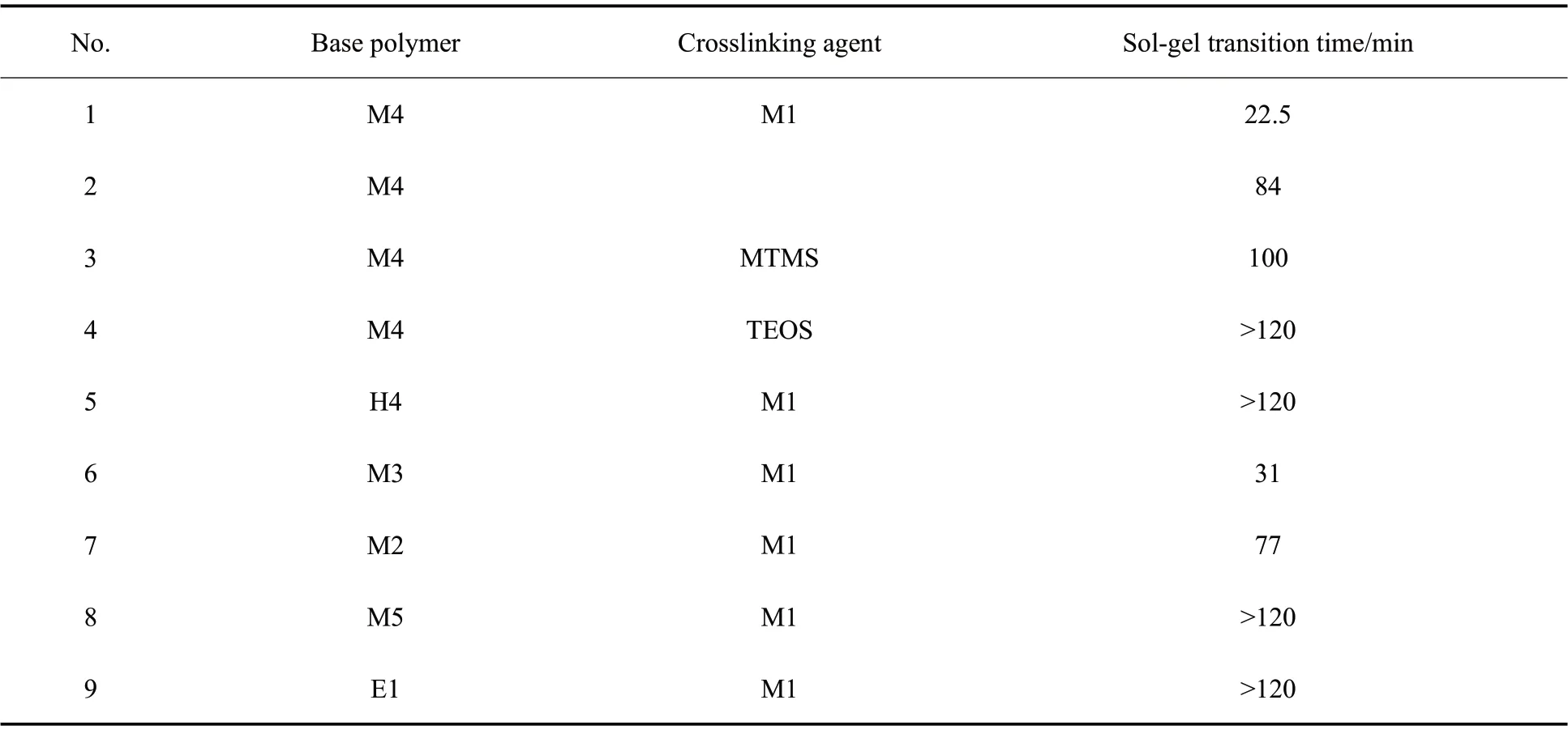
Tab. 3 Sol-gel transition time of different alkoxy-terminated poly(methyltrifluoropropyl)siloxane systems

Scheme 2 Shielding effect of trifluoropropyl on reactive endgroups

Tab. 2 GPC results of alkoxy-terminated poly(methyltrifluoropropyl)siloxanes (M1~M5, E1)and their precursors (H1~H4)a
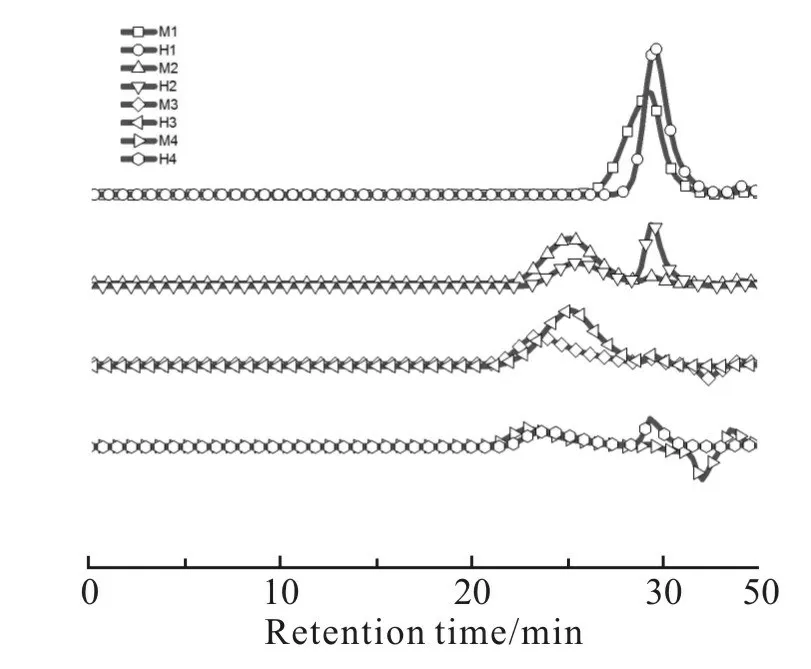
Fig. 1 GPC traces of alkoxy-terminated poly(methyltrifluoropropyl)siloxanes (M1~M4)and th eir precursors (H1~H4)
采用FT-IR 光谱和NMR 波谱对制备的聚合物进行了结构表征,测试结果与预期结构十分吻合。Fig. 2 给出了M1,E1 和H1 的FT-IR 光谱,可以看出,端羟基初始聚合物H1 在3316 cm-1处出现了Si—OH中羟基的伸缩振动峰,而此吸收峰在封端聚合物M1 和E1 中完全消失,这说明封端反应都很彻底,含氢硅氧烷对初始聚合物具有很好的封端效果。此 外,M1 光 谱 图 在2850 cm-1处 新 出 现 了SiOCH3中甲基的特征峰,而E1 光谱图在2930 cm-1处出现了SiOCH2CH3中—CH2—的吸收峰,这都证明了烷氧基封端的成功。

Fig. 2 FT-IR spectra of alkoxy-terminated poly(methyltrifluoropropyl)siloxanes (M1 and E1)and their precursor polymer (H1)
Fig. 3 给出了三甲氧基硅烷封端聚合物(M1,M2,M3 和M4)在氘代丙酮中的1H-NMR 和13C-NMR波谱图。可以看出,氘代丙酮溶剂在氢谱中的吸收峰 在δ2.04~2.07,在 碳 谱 中 的 吸 收 峰 在δ204.94,δ28.21~29.37 处,溶剂峰由*标出,溶剂中残余H2O 在氢谱中的吸收峰在δ2.8~2.9 处,由*标出。从核磁氢谱(Fig. 3A)中可以看出,与初始聚合物相同,三甲氧基硅烷封端聚合物在δ2.22~2.30,δ0.85~0.92 和δ0.25~0.32 处出现明显的共振吸收峰,分别对应于甲基三氟丙基硅氧烷结构单元中的SiCH2CH2CF3,SiCH2CH2CF3和SiCH3的化学位移。此外,封端聚合物在δ3.55~3.62 处出现新的吸收峰,此峰在较低相对分子质量聚合物M1 中最为明显,这对应于聚合物中新生成的端基SiOCH3。而在核磁碳谱(Fig. 3B)中,初始聚合物和封端聚合物都在δ123.84~132.06,δ26.82~27.79,δ8.21~8.74 和δ(-2.35)~(-1.79)处出现吸收峰,分别对应于结构单元中的SiCH2CH2CF3,SiCH2CH2CF3,SiCH2CH2CF3和SiCH3。封端聚合物中新出现的SiOCH3的化学位移出现在δ50.21~50.28处,此吸收峰在低分子量聚合物M1 的波谱中最为明显,原因是M1 中封端基的相对含量最高。

Fig. 31H-NMR (A) and13C-NMR (B) spectra of trimethoxy-terminated poly(methyltrifluoropropyl )siloxanes (M1,M2,M3 and M4) in acetone-D6
Fig. 4 给出了二甲氧基硅烷封端聚合物(M5)在氘代丙酮中的1H-NMR 和13C-NMR 波谱图。在核磁氢 谱(Fig. 4A)中,δ2.19~2.32,δ0.77~0.94 和δ0.25~0.32 处的吸收峰分别对应于结构单元中的SiCH2CH2CF3,SiCH2CH2CF3和SiCH3,δ0.15~0.19 和δ3.50~3.54 处的吸收峰对应于端基中新出现的SiCH3和SiOCH3。 在 核 磁 碳 谱(Fig. 4B)中,δ124.85~133.07,δ27.54~28.94,δ9.34~9.73 和δ(-1.77)~(-1.05)处的吸收峰分别对应于结构单元中的SiCH2CH2CF3,SiCH2CH2CF3,SiCH2CH2CF3和SiCH3,δ(-3.06)~(-2.74)和δ50.07~50.16 处的吸收峰对应于端基中的SiCH3和SiOCH3。三乙氧基封端聚合物(E1)的核磁表征结果与此类似。

Fig. 41H-NMR (A) and13C-NMR (B) spectra of methyldimethoxy-terminated poly(methyltrifluoropropyl)siloxanes (M5) in acetone-D6
2.2 室温交联流变性
M4 的 初 始 聚 合 物 的 黏 度 为2×104mPa· s 左右,而M1,M2,M3 的初始聚合物的黏度则分别为1×104mPa·s,5×103mPa·s 及100 mPa·s 左右,取初始聚合物黏度最高的M4 作为氟硅密封胶的基础聚合物。在氟硅密封胶的交联硫化过程中,体系黏度会不断升高直至转变为凝胶,因此可通过分析黏度增长趋势来研究交联反应的过程[6~8]。分别以MTMS,TEOS,M1 等硅氧烷为脱醇型交联剂,在二月桂酸二丁基锡催化下,体系黏度与交联反应时间的关系如Fig. 5 所示。

Fig. 5 Viscosity plotted against time during the crosslinking reaction of M4 as base polymer
可以看出,即使不加任何交联剂,M4 的黏度在有机锡催化下也出现明显的增长,这说明聚合物分子链两端的三甲氧基硅烷发生了自交联反应,与封端107 液体硅橡胶和硅烷封端聚醚(MS 聚合物)相同[9]。此外,当往M4 中加入4 份的M1 作交联剂后,黏度曲线的上升趋势显著加快,是已研究体系中最快的,这可从两方面进行理解。一方面,低分子量聚合物M1 的加入使体系中甲氧基浓度增加,因而极大提升了交联反应速率;另一方面,与MTMS,TEOS 等交联剂不同,M1 同样具有甲基三氟丙基硅氧主链结构,因而与基础聚合物M4 的相容性最好,于是表现出最高的交联反应速率。尤其值得注意的是,M4 表现出比其前驱体聚合物H4 更高的交联反应速率,可以明显地看出,同样采用M1 作交联剂时,H4 的黏度在测试时间2 h 内几乎没有变化。原因在于硅原子上的大体积三氟丙基侧基,其具有大的空间位阻,对H4 中的反应性端硅羟基具有很大的屏蔽作用,极大地影响了反应活性。而采用三甲氧基硅烷封端后,M4 中的反应性端甲氧基与三氟丙基的距离增大,因而屏蔽作用显著降低,而且反应性端基由1 个硅羟基转变为3 个甲氧基,官能度增加了3 倍,因而交联反应活性和速率明显提高,如Scheme 2 所示。
通过动态振荡实验进一步测试了M4 交联体系的储能模量(G′)和损耗模量(G″)随时间的变化,如Fig. 6 所示。曲线交点处G′=G″,弹性和黏性相等,处于溶胶-凝胶转变点,此凝胶点时间可作为交联反应速率的量度[8]。由Tab. 3 可以看出,M4-M1 体系达到溶胶-凝胶转变点的时间为22.5 min,比纯M4 的84min 和M4-MTMS 体系的100 min 都要早,这与黏度上升趋势是一致的,进一步证实了烷氧基硅烷的封端反应对聚合物交联反应活性的提升作用。在H4-M1 和M4-TEOS 体系的模量曲线中没有看到溶胶-凝胶转变点,这是因为交联反应速率太慢,凝胶未能出现在测试时间2 h 之内。此外,M3-M1 和M2-M1 体系的凝胶点时间分别为31 min 和77 min,说明基础聚合物的相对分子质量对交联速率的影响是有限的。而M5 和E1 为基础聚合物时,凝胶点时间都长于2 h,说明二甲氧基和三乙氧基封端聚合物的交联速率要低于三甲氧基封端聚合物。
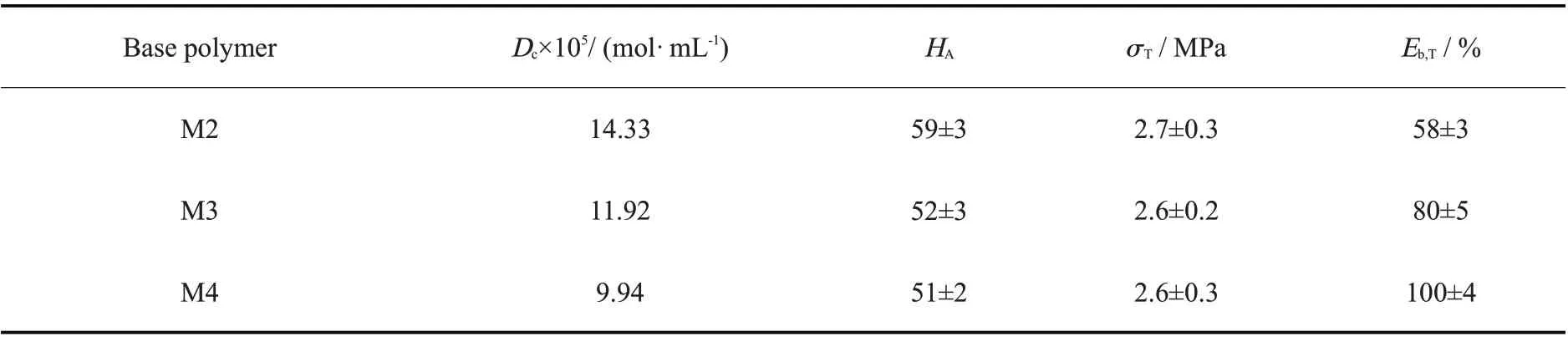
Tab. 4 Hardness and tensile properties of M2, M3 and M4 vulcanizatesa

Fig. 6 Storage modulus (G′) and loss modulus (G″)versus time during crosslinking reaction of M4 as base polymer
2.3 交联密度和力学性能
以M2~M4 为基础聚合物,向其中加入35 phr 气相二氧化硅进行增强,以M1 和有机锡为交联剂和催化剂,设计制备脱醇型氟硅密封胶,在温度23 ℃、相对湿度50%条件下硫化24 h 后,通过低场核磁法测试其交联密度,结果如Tab. 4。可以看出,基础聚合物为M2,M3 和M4 的交联密度分别为14.33×10-5mol/mL,11.92×10-5mol/mL 和9.94×10-5mol/mL,这说明3 种体系都发生了交联反应,并且单位体积内的交联点数量会随着基础聚合物相对分子质量的增大而呈现降低趋势。
进一步测试了3 种密封胶体系的力学性能,结果列于Tab.4。可以看出,在气相二氧化硅增强下,密封胶的邵氏A 硬度达到50 以上,拉伸强度为2.6 MPa,断裂伸长率为50%~100%。
2.4 热稳定性和玻璃化转变温度
通过TGA 研究了3 种氟硅密封胶的热稳定性,结果如Fig.7 所示。由TGA 曲线可以看出,基础聚合物为M2,M3 和M4 的硫化胶具有较高的初始分解温度(T5d),分别为430 ℃,400 ℃和373℃,这是因为氟硅聚合物主链本身的热稳定性优异,Si—O 键具有较高的键能(466 kJ/mol),同时氧化铁红作为耐热添加剂又起到抑制自由基增长、延缓聚合物分解的作用[10]。从DTG 曲线还可看出,3 种硫化胶的最大热分解速率为1.44%~1.58%℃-1,最快热分解的温度均高于500 ℃。

Fig. 7 TGA and DTG curves of M2, M3 and M4 vulcanizates

Fig. 8 DSC (a) curves and FT-IR spectra (b) of M2, M3 and M4 vulcanizates
Fig.8(a)给出了3 种硫化胶的DSC 曲线。M2,M3 和M4 硫化胶的玻璃化转变温度(Tg)分别为-69.3 ℃,-67.4 ℃和-68.9 ℃,可以看出,M3 的Tg是3 组样品里最高的,这是因为3 组样品都产生了分子间氢键(Fig. 8(b)),M3 的氢键密度大于M4 大于M2,Tg随分子间氢键密度增大而升高[11],因此M3的Tg最高。虽然3 组样品的Tg因分子间结构的差异而略有不同,但均与传统的氟硅橡胶在同一水平,具有较好的耐低温性能[12]。
3 结论
(1)以4 种黏度不同的α,ω-端羟基聚甲基三氟丙基硅氧烷为初始聚合物,通过与烷氧基硅烷的脱氢缩合封端反应,合成了6 种烷氧基封端氟硅聚合物——三甲氧基硅烷封端聚合物(M1~M4)、二甲氧基硅烷封端聚合物(M5)和三乙氧基硅烷封端聚合物(E1)。通过FT-IR 光谱和NMR 波谱对所得聚合物进行了结构表征,证实了预期结构的生成。
(2)通过流变仪研究了烷氧基封端氟硅聚合物在室温交联硫化过程中的流变学,采用稳态旋转模式测定了体系的黏度,采用动态振荡模式测定了体系的储能模量(G′)和损耗模量(G″)。结果表明,与初始聚合物(H4)相比,封端聚合物(M4)的黏度上升更快,凝胶点时间更短,这说明M4 具有更高的交联反应速率。究其原因,在初始聚合物中,与端羟基相连的硅原子直接连着大体积的三氟丙基侧基,端羟基受到三氟丙基的屏蔽作用,因此反应性很低;而在封端聚合物中,反应性烷氧基与三氟丙基侧基的距离增大,屏蔽作用大大降低,因此交联反应性提高。此外,烷氧基封端聚合物(如M4)还具有自交联性能,而低分子量的M1 可作为交联剂,具有比甲基二甲氧基硅烷(MDMS)和正硅酸乙酯(TEOS)更快的交联速度,原因是前者与基础聚合物具有更好的相容性。
(3)以M2~M4 为基础聚合物制备了氟硅密封胶,其具有较好的交联密度和力学性能,热稳定性和玻璃化转变温度也在典型的氟硅聚合物范围内。
