人工智能辅助的蛋白质工程
2022-07-15卞佳豪杨广宇
卞佳豪,杨广宇
(上海交通大学 生命科学技术学院,微生物代谢国家重点实验室,上海 200240)
合成生物学是一个广泛的研究领域,通过将生物学和工程学相结合来设计和创建具有新颖功能的生物系统[1-2]。这一过程需要功能各异、形式多样并且能够良好实现预期功能的生物元件,特别是蛋白质功能元件(催化酶、转录因子、转运蛋白、蛋白支架等)[3]。但是,天然来源蛋白质元件大部分都不能满足人工生物系统的需要,实际应用中往往表现出折叠错误、细胞毒性、功能不适宜等缺陷[4-5]。蛋白质从头设计或对天然蛋白质进行分子改造,成为解决这一问题的重要途径。对于蛋白质或酶的分子改造,已经成为合成生物学的重要研究领域[6-9]。
在天然蛋白质分子改造方面,主要包括定向进化(directed evolution)和理性设计(rational design)两种策略[10-12],见图1。前者通过模拟自然选择过程,对目标基因进行多轮突变和筛选实验,直至获得所需水平的优良变体,但是该技术受限制于较低的筛选速率和序列空间中庞大的变体数量[13]。后者依据序列和结构信息,选择较少的关键位点进行精准改造,从而构建较小的突变文库,但是需要对结构功能信息有深入了解,并且需要巨大的计算资源[14]。

图1 理性设计,定向进化和人工智能辅助的蛋白质工程策略示意图(理性设计依赖序列和结构信息,精准设计突变体文库,但难以应用于缺少结构功能信息的蛋白质。定向进化中对目标基因进行多轮突变和筛选实验,不受结构功能信息限制,但是需要进行高通量的筛选方法。人工智能辅助的蛋白质工程则需要大量的序列-功能数据,可以来源于实验、计算和数据库等多方面,通过构建的预测模型,能够更有效地探索蛋白质突变体序列空间)Fig.1 Schematic diagram for rational design,directed evolution and artificial intelligence-assisted protein engineering(Rational design relies on sequence and structural information to design mutant libraries accurately.However, it is difficult for being applied to pro‐teins lacking structural and functional information.In the directed evolution strategy,multiple rounds of mutation and screening experiments are per‐formed on target genes,which are not limited by structural and functional information,but high-throughput screening methods are required.Artificial intelligence-assisted protein engineering requires a large amount of sequence-function data,which can be derived from experiments,calculations,and databases.Through the predictive model,the sequence space of protein mutants can be explored more effectively)
人工智能辅助的蛋白质工程策略是一种由数据驱动的新策略[15]。该策略通过学习已有数据中的信息,建立起输入属性(如序列)到输出属性(如功能)的映射关系,不需要详细的物理或生物层面的基础信息[16]。一旦得到足够准确的映射关系(或者说预测模型),就能够通过实验中容易得到的输入值来预测输出值,从而免除大量的重复性实验。目前,该策略已经成功应用在蛋白质工程的很多方面,包括蛋白分子结构预测[17-18]、蛋白分子功能预测[19-20]、蛋白分子溶解度预测[21-22]和指导设计智能组合文库[23-26]等。
目前已有多篇综述详细介绍了有关机器学习的基础概念[27-31]。这些文章多从数据和算法的角度来对人工智能的主要进展进行了介绍,但是对于非生物信息学背景的研究人员而言,这类综述读起来较为深奥。为了使更多实验生物学背景的人员理解人工智能蛋白设计的进展,本文将主要介绍人工智能辅助蛋白分子设计的应用实例、已开发的数据库和平台工具等几个方面,为希望进入人工智能蛋白质工程领域的入门者提供帮助。
1 人工智能辅助的蛋白质工程应用实例
人工智能算法由于准确度高、计算速度快、不受蛋白质结构功能信息限制等优点,近年来被大量应用于蛋白质工程领域,包括蛋白质的结构、功能、热稳定性、对映体选择性、光敏性及指导设计智能组合文库等多个方面。其中除了经典的机器学习算法(决策树、支持向量机和高斯过程回归等)外,多种深度学习算法和基于深度学习的自然语言处理技术也获得了成功的应用。在下文中,我们重点集中于近几年在蛋白质结构预测、功能预测、溶解度预测和指导设计智能组合文库四个方面的成功案例,系统地分析人工智能算法在蛋白质工程中应用的优势。
1.1 蛋白质结构预测
截至2018 年,蛋白质数据库中发布了超过145 000 个蛋白质结构,但与目前已知的超过2 亿条蛋白质序列相比,仍仅占很小的比例[32],因此蛋白质结构预测是生物学中经久不衰的热点问题。早在1992 年,机器学习算法就被用于预测蛋白质二级结构[33]。近几年,利用深度学习算法和蛋白质序列的三维结构预测模型取得了不小的进展[34]。首先是在2018 年第13 届全球蛋白质结构预测竞赛(CASP)上,AlphaFold 模型结合深度残差卷积神经网络和快速Rosetta 模型,获得了预测43 种蛋白中的25 种蛋白结构的最高分,实现了预测成功率的 突 破[17]。2019 年 底,David Baker 团 队 发 表 了trRosetta 方案,综合了深度学习和Rosetta 的优势和进展,具有良好预测精度的同时,能够在本地电脑上就可以完成计算,使得预测蛋白结构的门槛 大 大 降 低[18]。 在2020 年 的CASP14 中,AlphaFold 2 再次获得冠军。根据DeepMind 官方的信息,AlphaFold 2 在无模板的自由建模任务中,拿 到 了87.0 的GDT_TS 分 数(global distance test[35]),在常规项目中拿到了92.4 分,这意味着该系统预测的均方根偏差(即预测数据与实验数据在原子位置上的偏差)大约为0.16 nm,已经达到了常规蛋白质晶体结构的实验精度。尽管AlphaFold 目前最好的成绩是针对单链蛋白质分子,但这种成绩本身就足以证明人工智能算法在蛋白质结构预测中的巨大潜力,例如减少繁琐的结晶条件探索工作,以及提供以常规实验方法难以获得的蛋白质结构等。
1.2 蛋白质功能预测
天然蛋白的功能表征实验需要大量工作,其速度远远低于新蛋白序列的获取速度[36-37]。借助人工智能算法来预测蛋白质的功能是另外一个研究热点。2018 年,研究者通过收集来自拟南芥的54 种GT1 家族糖基转移酶的序列信息和它们91 种底物的物理化学特性(如疏水常数lgP、分子表面积)和结构信息(如官能团拷贝数、框架类型),建立了初始的数据集,并以多种基于决策树的算法来构建酶功能的预测模型(图2)[19]。在不需要进行任何实验的条件下,该预测模型利用酶序列,就能够准确地预测其他植物中(苜蓿和燕麦)GT1糖基转移酶的活性,对来自细菌的GT1 酶活性的预测准确率也在70%以上。这表明能够利用高通量数据进行学习的人工智能算法在底物混杂、已解析结构少的酶的功能注释中具有巨大潜力。此外,人工智能算法也被应用于预测酶的EC 编号(enzyme commission number),帮助对酶分子进行分 类。先 后 发 展 出 的PRIAM[38]、CatFam[39]、EFICAz2.5[40]、 SVM-prot[41]、 COFACTOR[42]、DEEPre[36]、 DETECT v2[43]、 ECPred[44]和DeepEC[20]等多种预测工具,在计算时间、计算精度和覆盖范围等预测性能方面逐渐改进,简要内容见表1。其中,DeepEC 方法包括三个独立的卷积神经网络,利用氨基酸序列,就能对氨基酸序列是否为酶分子、酶分子EC 编号的三位和四位数值进行预测。与CatFam、DETECT v2、ECPred、EFICAz2.5 和PRIAM 五种代表性的酶EC 编号预测工具相比,在Swiss-Prot 数据库中选取的201 个酶进行验证时, DeepEC 表现最佳, 准确率(accuracy)和召回率(recall)分别为0.920和0.455。即45.5%的阳性样本能被预测模型准确识别,这其中92.0%样本的预测值与真实值是一致的。

表1 EC编号预测工具汇总表Tab.1 Forecast tools for EC numbers

图2 GT1家族糖基转移酶预测模型(GT-Predict)的工作流程[19](基于功能的算法学习方法GT-Predict,使用来源于酶、亲电试剂和亲核试剂的多种训练集来创建基于物理化学和局部序列的分类器,从而预测GT1糖基转移酶的催化活性和功能信息。Nuc表示亲核基团的数量/类型)Fig.2 Workflow for predicting the GT1 glycosyltransferase model(GT-Predict)[19](The function-based algorithmic learning approach,GT-Predict,uses a diverse training set of enzymes,electrophiles,and nucleophiles to create a physicochemical and local-sequence-based classifier for predicting the novel transformations and functional annotation of GT group-transfer enzymes.)
1.3 蛋白质溶解度预测
蛋白质的溶解度对于其行使功能起到重要作用。溶解度过低是蛋白质大规模生产中常见的主要瓶颈[45-46],而溶解度的测量费时费力,因此非常需要能够准确对蛋白质溶解度进行预测的生物信息学工具。新加坡国立大学的Han 等[21]测试了逻辑回归、决策树、支持向量机、朴素贝叶斯、条件随机森林、XGboost和人工神经网络等七种算法构建基于序列的溶解度预测模型,其中支持向量机算法构建的模型在此预测任务中显示出最高的准确性。在预测结果为代表“可溶”和“不溶”的二分值“1”和“0”时,该模型的预测准确率为0.7628。除此之外,该模型还可以预测蛋白质连续的溶解度值(离心后上清液的蛋白质质量与总蛋白质质量之比)。但这种情况下,模型预测的准确性有所降低,决定系数为0.41。最近,中山大学的Chen Jianwen 等[22]利用蛋白质接触图(contact map)和图神经网络算法(GCN)开发了一种新的利用氨基酸序列预测蛋白质溶解的模型GraphSol,在同样利用eSOL 数据库中的蛋白质溶解度数据进行验证时,进一步提升了预测模型的性能,其决定系数为0.48。在蛋白质工程中,输出结果为简单的二分值时,重要的氨基酸突变对溶解度的贡献无法分析。例如,“不溶”和“可溶”的群体中,不同突变对蛋白质溶解度的贡献无法分辨。并且,当存在大量“可溶”的预测变体时,无法从中选出表现最佳的少数变体进行实验验证[21]。因此,能够预测蛋白质连续的溶解度的模型更适用于辅助蛋白质工程。随着可用数据集的扩大和算法框架的优化,基于序列的蛋白质溶解度预测模型将能够有越来越高的准确率。
1.4 指导设计智能组合文库
人工智能策略在酶定向进化中也具有重要的应用潜力。依靠人工智能算法,可以基于已有的序列/结构信息,直接建立起序列/结构-功能的映射关系,因此理论上可以极大减少筛选工作量,并且更加有效地探索整个组合突变体的序列空间[26,47]。例如,在指导绿色荧光蛋白向黄色荧光蛋白进化的研究中,研究者们对选定的四个关键位点构建了单点饱和突变库和随机诱变库,共包含218 个变体。但将所有变体筛选之后,没有发现比参考黄色荧光蛋白性能更好的突变体。随后,他们选择其中的155 个变体的序列-功能数据作为初始数据集,以高斯过程回归算法来构建预测模型。通过预测模型,遍历了整个四点组合序列空间中的近16 万个变体,并对其性能打分。在仅仅对预测突变体文库中排名靠前的78 个变体进行验证的情况下,就找到了12 个黄色荧光强度高于参考蛋白的突变体[23]。
此外,在Frances H.Arnold 团队[24]的研究中,他们从对S-对映体有76%ee 一氧化氮双加氧酶出发,利用455个突变体来构建从序列预测功能的模型。通过该模型对涵盖了七个位置(两个区域)的组合序列空间中约168 000 个变体的性能进行预测,再进行两轮筛选,共验证了360个变体后,就获得了对S-对映体有93%ee和对R-对映体有79%ee的两种优良变体。
在2018 年,Manfred T.Reetz 团队[25]利用一种innov’SAR 的人工智能方法来指导在环氧水解酶的对映体选择性的进化过程中组合突变文库的设计,在仅使用了38 个突变体的序列-功能数据的情况下,预测模型对九个位点上共512 种突变体的功能进行了预测,经过简单验证后就找到了多个优于经随机突变文库筛选得到的最佳突变体的酶分子。
2019年,为了解决视紫红质通道蛋白筛选通量太低,并且要同时保留其多种特性的问题,Frances H.Arnold团队[26]使用了人工智能辅助的蛋白质工程策略(图3)。其方法为首先利用实验表征的和文献报道得到的183个序列-功能数据,构建一个分类模型,从而有效排除重组文库120 000 条序列中绝大多数的非功能序列。然后根据已经表征的视紫红质通道蛋白的特性信息,针对不同的目标属性来建立不同的回归模型,例如电流强度、关闭动力学(即曝光后通道关闭所需的时间)和激活的波长敏感度等,对所有具有功能的序列进行特性的得分的预测。最后从预测库中选择少部分排名靠前的突变体(28个)进行实验验证,并得到了目标属性都优于现有的视紫红质通道蛋白的三个变体ChRger1、ChRger2和ChRger3。

图3 人工智能辅助的视紫红质通道蛋白改造的工作流程[26][在重组文库中表征的102种ChR蛋白和文献中报道的61种变体,共同构成了(1)分类模型的训练集。然后,使用经过训练的分类模型来预测12000个未表征的ChR序列变体是否具有功能。接下来,构建了三个(2)回归模型,分别针对不同的ChR光电流特性:光电流强度,关闭动力学和光电流的波长敏感性]Fig.3 Workflow for machine learning-guided channelrhodopsin engineering[26][102 ChR proteins characterized in the recombinant library,together with 61 variants reported in the literature,constitute the training set of theclassification model(1).Then the trained classification model was used to predict whether 12000 uncharacterized ChR sequence variants are functional,and three regression models(2)were trained,one for each of the ChR photocurrent properties of interest:photocurrent strength,off-kinetics and wavelength sensitivity of the photocurrents.]
2 人工智能辅助的蛋白分子设计策略概述
在人工智能辅助的蛋白分子设计策略中,本质是基于已有的数据,引入不同的机器学习算法来进行“输入特征-输出特征”的映射关系的构建。根据训练数据是否拥有标记信息(即规定的输出值),机器学习大致可划分为监督学习(supervised learning)和无监督学习(unsupervised learning)。由于在蛋白质工程中,最终目的是获得或者优化目标蛋白的一个或多个属性,因此至少会有一个属性值作为标记信息,属于监督学习[48]。
图4描述了监督学习的工作流程,主要可以分为三个步骤[27]。步骤1: 通过数据库、实验和文献等方式收集初始数据,将序列作为输入特征,将蛋白质的功能信息(如对某种目标底物的活性)作为标记信息(如1 代表该序列对底物有活性,0 代表该序列无活性),转为计算机能够识别的数字格式,并拆分为训练集和测试集。步骤2:选用合适的算法,利用训练集进行预测模型的训练,建立起“序列-活性”的映射关系。步骤3:利用训练的模型,输入测试集的序列,得到预测值(0或1),通过比较测试集中的真实值和预测值之间的差异,评估预测模型的性能。在整个流程中,有两个关键点对预测模型的性能至关重要:数据、分子描述符和算法,人工智能方法的开发重点也是集中于这两个方面。
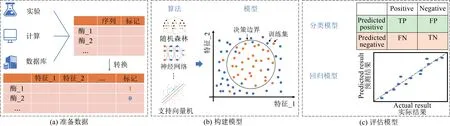
图4 监督学习的流程示意图[27](a)准备数据:来源于实验,计算或数据库的数据通常会转换成计算机可以识别的格式,并拆分为训练集和测试集;(b)构建预测模型:利用训练集训练不同的算法以找到决策边界,构建预测模型,例如随机森林,神经网络和支持向量机;(c)验证模型:对于分类问题或者回归问题,应选择合适的评估方法Fig.4 Schematic diagram of the supervised learning process[27]Step(a):Preparing data.The data from experiments,calculations or databases are usually converted to a format that the computer can recognize and split into the training and test parts.Step(b):Constructing a predictive model.Using the training set to train different algorithms to find decision boundaries,such as random forests,neural networks and support vector machines,so as to build predictive models.Step(c):Validating the model.An appropriate evaluation method should be selected for tasks with classification or regression.
2.1 数据
由于人工智能算法严重依赖数据,初始数据的数量和质量决定了训练得到的模型的泛化性能[49-50]。数据集的数量不足或者质量过低会导致模型出现过拟合或者欠拟合的问题,往往会进行交叉验证来检测模型中是否存在该问题,例如k折交叉验证(即将整个数据集平均拆分为k份,每一份轮流作为测试集,其余作为训练集,如图5),因此数据收集是重要且耗时的步骤。一般来说,人工智能辅助策略很适合与其他蛋白质改造策略联用,利用在随机突变或(半)理性设计后生成的数据作为初始数据[51]。但是,一方面,就来自单轮实验的数据而言,数据集通常仅包括数十种到数百种变体,这在人工智能算法框架中属于较小的样本量[52]。另一方面,从实验中以及部分数据库中的数据是存在一定偏差的,特别是针对蛋白质某项属性进行改造时,表现不好的突变体通常直接被丢弃掉,因此导致初始数据集中数据不均匀。因此,如果采用人工智能辅助的蛋白质工程策略,应当注意收集阴性数据来保证数据的无偏性。针对训练数据的数量偏少的问题,一方面许多数据库一直在收集、整理来源于文献或实验的数据,涵盖蛋白质的序列、结构、功能和溶解度等多个属性,可以为人工智能算法提供许多优质的数据;另一方面,随着超高通量筛选和二代测序等高通量生物学实验技术的逐渐成熟,可以相信在不远的未来可用数据的数量和质量都会得到大幅度的提升,为更精准的人工智能算法提供充足的资源。
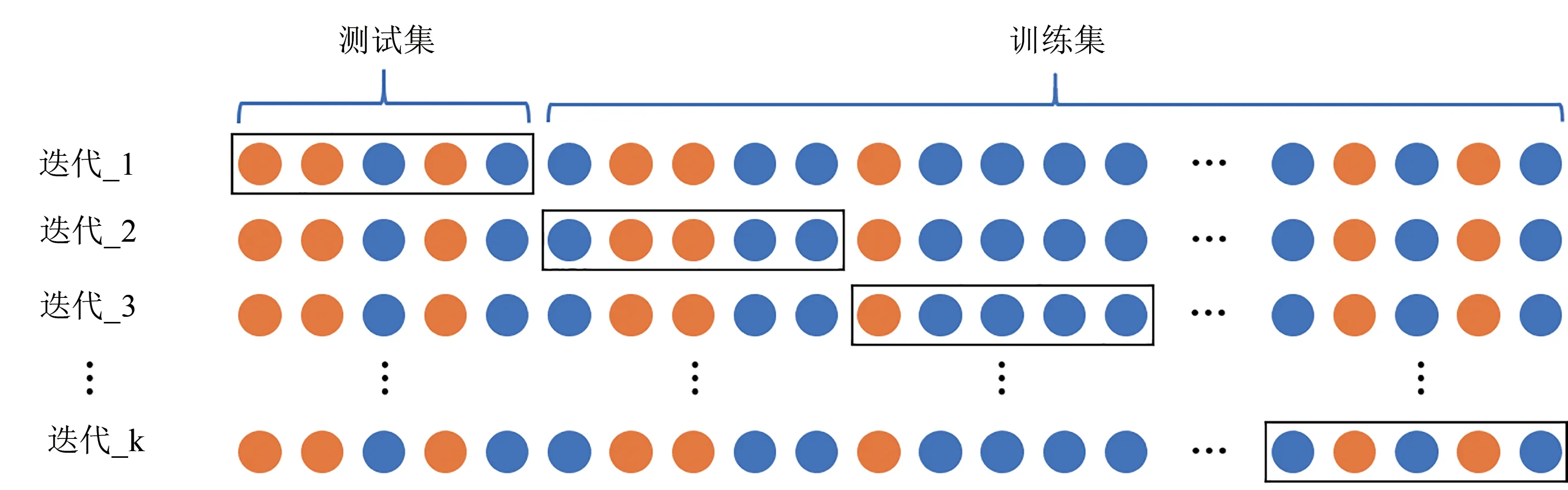
图5 k折交叉验证示意图(将训练数据进一步细分为k个子集,并且将训练工作流程重复k次,同时保留k个子集中的一个用于评估,其余k-1个子集用于训练)Fig.5 Schematic diagram for k-fold cross-validation(The training data is further subsplit into k subsets,and the training workflow is repeated k times with each of the k subsets holding for evaluation and the remaining k-1 subsets used for training)
2.2 分子描述符
分子描述符(molecular descriptors),就是将分子的化学信息(例如结构特征)转换成有用的数字形式的工具。算法,即学习算法(learning algorithm),是机器学习中用于帮助计算机系统从数据中产生模型(model)、总结“经验”的方法[53]。但计算机系统仅能理解数字向量,所以算法不能直接作用于蛋白质序列[16]。因此,在获得序列之后,一般还需要利用合适的分子描述符将氨基酸序列处理为计算机能够识别的格式。以最简单的独热编码描述符为例,对于N个长度为L的多个蛋白质突变体序列,它们若在某一相同位点上包含S种不同的氨基酸(S≤N,S≤20),则该位置的所有氨基酸都可以用一个S维向量表示,每一个S维向量都包括S−1 个0 和一个1,其中1 的位置表明该氨基酸的身份,如图6。氨基酸序列也可以根据物理性质进行编码,每种氨基酸可以由其电荷、体积或疏水性等特性或者这些特性的组合来表示,如AAindex[54]中就包含了大量类似的描述符。目前常用到的描述符有4 种类型,包括基于氨基酸序列特征的描述符、结构信息描述符、嵌入式表示描述符以及突变指示描述符,在综述[16,30,55]中均有详细描述,本文不再赘述。
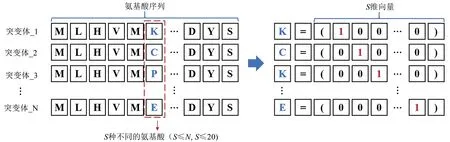
图6 独热编码示意图(N个蛋白质突变体序列中L个氨基酸中某一相同位置包含S种不同的氨基酸,独热编码将这S个氨基酸都表示为包括S-1个0和一个1的S维向量,其中1的位置表示该位置的氨基酸的种类)Fig.6 Schematic diagram for one-hot encoding(A certain position of the L amino acids in the N protein mutant sequence contains S different amino acids.The one-hot encoding represents all S amino acids as an S-dimensional vector including S-1 zeros and one 1.The position of 1 indicates the type of amino acid at that position.)
2.3 算法
除此之外,人工智能领域也已经提出了大量算法。从模型复杂度角度, 机器学习分为经典机器学习和深度学习[56]。前者中的偏最小二乘回归[57]、支持向量机[58]、决策树/随机森林[59]和贝叶斯网络[60]等常见算法以及后者中的变分自编码器[61]、卷积神经网络[62]和循环神经网络[63]等都已用于辅助蛋白分子设计。
经典机器学习和深度学习二者的不同在于,经典机器学习算法强烈依赖于人工提取的特征,一般与基于氨基酸特征或序列整体特征的分子描述符配套使用,但可能会受限于定义好的特征值而忽略数据中隐藏的信息[64]。而深度学习是通过深度神经网络,将数据进行分层抽象处理,能有效排除噪声、发现隐藏信息,因此非常适用于从高维数据发现复杂结构[56]。各个算法的入门介绍可以参考综述[16,28,31,55]。
在选择算法时,一般会以线性模型作为基线。如果线性模型的准确性不足,并且初始数据集中数据小于10 000 时,偏最小二乘回归、随机森林和支持向量机都可能构建出最佳的预测模型,而神经网络则通常在更大的数据集上表现出最佳性能[16]。在计算速度方面,由于复杂程度和所需训练集大小等因素影响,深度学习往往也需要花费更多时间[55]。因此,如何选择合适的算法,需要研究者在具体的预测任务中仔细衡量准确率、计算速度和实现难度等因素。
在人工智能辅助的酶定向进化策略中,选择合适的分子描述符和机器学习算法对构建准确的预测模型而言至关重要。没有一种分子描述符和算法能够满足所有的学习任务[65],研究人员必须结合专业知识或者同时构建多个模型进行比较。Frances H.Arnold 团队使用高斯过程算法,嵌入式表示、蛋白质指数和独热编码等氨基酸编码方式进行了未知功能蛋白的功能预测,结果发现,使用嵌入式表示描述符训练的模型预测能力与其他模型的预测能力相当,甚至超过它们[66];而在Jennifer M.Johnston 等人的研究中,使用多种描述符和卷积神经网络模型构建了蛋白质序列/活性关系预测模型,结果发现,基于序列的氨基酸特性相关描述符的卷积神经网络模型表现较好,而嵌入式表示描述符表现不佳[55]。这恰恰证明了没有一种分子描述符和算法能够满足所有的学习任务。
3 相关的数据库和线上平台
3.1 数据库
除了与其他分子改造策略联用之外,随着高通量筛选和二代测序技术的不断发展,越来越多的蛋白质信息被挖掘,目前已经有许多优秀的数据库收集并整理了多种可作为该策略初始数据的信息,是优良的数据来源。即便数据库中大量蛋白质序列信息没有功能注释,也可以用于构建预测模型,即通过人工智能算法从这些序列中学习、提取特征,然后作为下一步从“已知特征”到“目的属性”的顶层预测模型的输入数据。例如,在2019 年George M.Church 团队利用了大约2400 万条蛋白质序列训练递归神经网络算法,构建了一个UniRep 模型[67]。该模型能够预测氨基酸序列中下一个氨基酸是什么,以此来提取氨基酸序列中不可见的特征。这些特征可以作为其他算法(如随机森林、稀疏线性回归等)的输入信息,来构建顶层特征(图7)。在应用方面,基于UniRep 模型的预测模型在预测蛋白质稳定性和荧光蛋白序列优化任务中,性能都明显优于Frances H.Arnold 团队曾报道的Doc2Vec 模型[66]。该研究说明人工智能算法能够深度挖掘蛋白质序列中隐藏信息,为提高蛋白质工程的效率、解决蛋白质表征实验费时费力问题提供了一个全新的方法。

图7 UniRep模型的工作流程[67][在训练部分,UniRep模型使用了2400万个氨基酸序列作为训练集。然后使用训练好的模型来预测下一个氨基酸(使交叉熵损失最小化),从而学会如何正确表示氨基酸。在应用部分中,训练后的模型通过提取和平均各个氨基酸的数字向量,从而生成输入序列的单个固定长度矢量表示。这些向量可以用于训练顶级模型,从而应用于多种序列-功能预测任务]Fig.7 Workflow for the UniRep model[67][In the training part,24 million amino acid sequences are used to train the UniRep model.Then the trained model is used to predict the next amino acid(minimizing the cross-entropy loss),so as to learn how to correctly represent the amino acid.In the application part,by extracting and assessing the numerical vector associated with the amino acid,the trained model is used to generate a single fixed-length vector representing the input sequence.Next,these vectors can be used to train top models,which can be applied to various sequence-function prediction tasks.]
除了最常见的蛋白质序列和结构数据库外,越来越多的数据库在自动或手动收集整理蛋白质突变稳定性、溶解度等信息,表2对部分比较常见的数据库的类型、大小和特点进行了介绍。
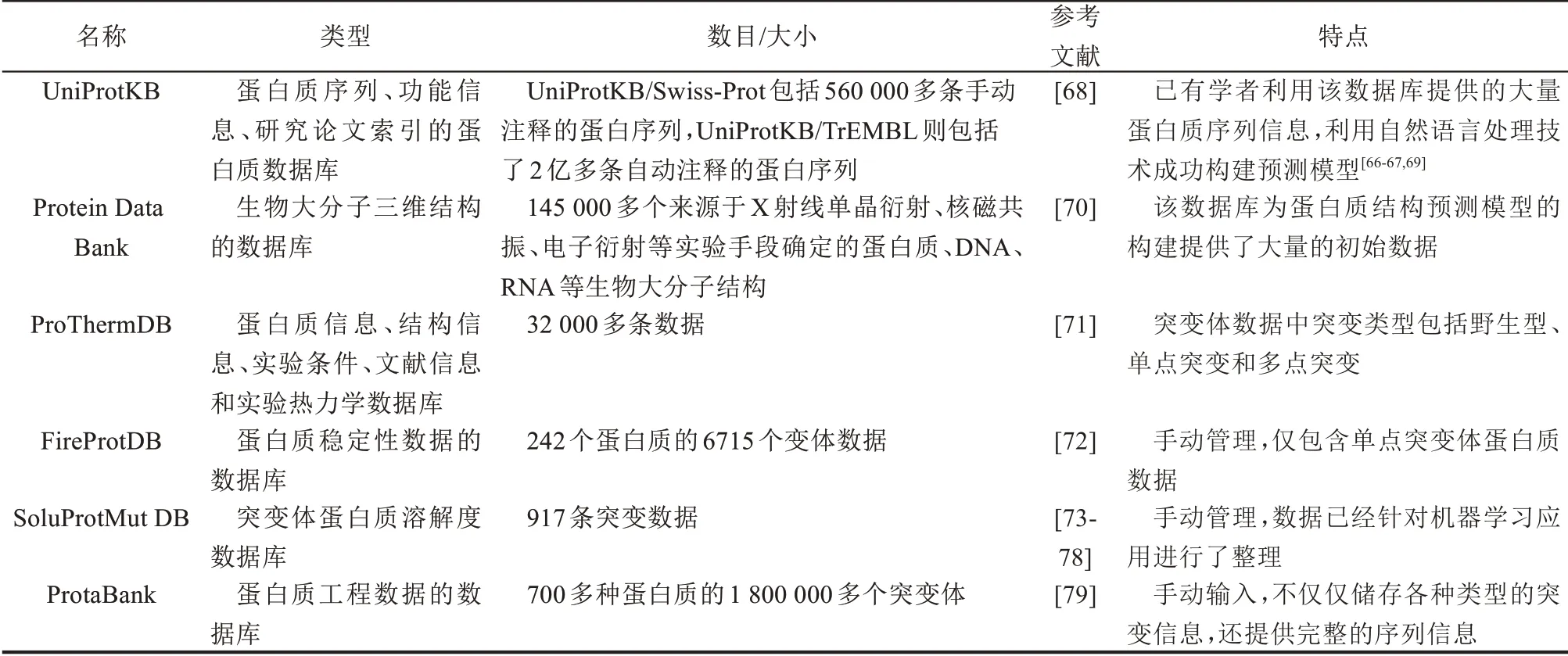
表2 常见数据库汇总表Tab.2 Commonly used database
3.2 线上平台
事实上,学者们已经开发了许多线上平台或者工具包来帮助人们获得蛋白质序列中的特征信息以及使用人工智能算法的工具,汇总信息见表3。大多数工具包和线上平台都只关注于完成整个生物序列分析任务的一部分,例如,大多数工具都只能利用不同类型的分子描述符从序列中生成特征。但是其中BioSeq-Analysis2.0 和iLearn 两个平台可以自动执行整个蛋白序列分析任务的步骤,SOLart 平台则额外引入了结构信息来预测目标蛋白质溶解度,下面进行详细阐述。

表3 基于蛋白质序列的特征生成工具汇总表Tab.3 Feature generation tools based on protein sequences
3.2.1 Protein-Analysis2.0
Protein-Analysis2.0 是服务器BioSeq-Analysis2.0的线上蛋白质服务器,可以通过三个主要步骤完成蛋白序列分析任务:特征提取,预测模型构建以及性能评估[91]。其中在特征提取方面,包括13 种基于氨基酸残基水平的分子描述符和39 种基于氨基酸序列水平的分子描述符。为了避免某些分子描述符导致编码后向量维度爆炸的情况,该平台还添加了两种特征选择方法。在人工智能算法方面,该平台仅整合两种分类算法(支持向量机和随机森林)和一种序列标记算法(条件随机场)。在性能评估方面,该平台支持5 折交叉验证或独立数据集两种方式。同时,作者利用文献[94]中的数据作为基准数据,预测蛋白质的无序区域,其中,其中条件随机场-One-hot(6-bit)预测模型表现最佳,与文献中的方法高度可比,证明了该平台的实用性。平台地址为:
http://bliulab.net/BioSeq-Analysis2.0/home/
3.2.2 iLearn
iLearn线上平台与BioSeq-Analysis2.0类似,不同之处在于:①iLearn平台中包含更多种分子描述;②拥有更丰富的特征分析功能,支持聚类、特征向量归一化、降维和5种特征选择方法;③支持更多的机器学习算法和更多的评估指标;④选择一种或多种机器学习算法进行提交,可以返回具有最佳性能的模型等[93]。在应用方面,作者从文献[95]中收集初始数据集和独立测试数据集,利用BLOSUM62、CKSAAP、Binary、Z-scales、AAindex、AAC 和EAAC其中不同的分子描述符来进行蛋白质丙二酰化位点预测模型的构建,最终EAAC编码模型的AUC值为0.73,与原始工作中报告的AUC 值为0.739 相当,表明iLearn可以作为一种方便有效的工具来构建相关的预测模型。平台地址为:
https://ilearn.erc.monash.edu/
3.2.3 SOLart
SOLart线上平台要求的输入信息仅仅是蛋白质结构,该结构可以由用户手动上传,也可以从Protein Data Bank 自动上传,无需其他额外操作。其原理是在基于序列的特征(如蛋白长度和氨基酸组成)之外,引入了溶解度依赖距离电位、溶剂可及表面积和二级结构等结构特征,并以此训练随机森林算法构建预测模型。在交叉验证中,实验和预测的溶解度值之间的皮尔森相关系数几乎达到0.7,表现出了较好的预测能力[93]。平台地址为:
http://babylone.ulb.ac.be/SOLART/index.php
4 总结
目前人工智能策略在蛋白质工程领域的应用范围主要包括蛋白质结构预测、酶功能预测、蛋白质溶解度预测以及指导智能组合文库设计等。在短短数年中,人工智能策略已经在蛋白质工程领域展现了显而易见的应用潜力和价值。要进一步挖掘人工智能在蛋白质工程领域的潜能,提升预测模型的性能,还需解决许多问题。首先,目前数据库中自动注释的蛋白质的信息质量难以让人信服,手动管理的高质量数据库中数据量的大小又远不如前者,缺少大量可用于训练和验证的标准化的数据。在后续工作中,应该构建更加高质量的基础性蛋白质序列-结构-功能数据库,有助于更加高效地构建人工智能预测模型。其数据集应该是相关的、有代表性的、非冗余的,并且包含通过实验确定的阳性和阴性数据,具有统一的标准格式等[50]。其次,在早期的实验中,更容易被表征或者具有更好表型的蛋白质往往会在后续工作中进行表征和确认,而表现不佳的蛋白质则会被丢弃,导致数据出现偏差,模型的预测性能下降[96]。此外,人工智能辅助的蛋白质工程策略还处于早期阶段,大多数例子中的预测模型可能无法直接推广应用到其他学习任务中,需要重新进行训练和验证。最后,随着越来越多的复杂的人工智能算法被用于蛋白质工程,难以对预测模型的原理进行解释等等。
随着相关研究的逐渐深入,最近已经有一些针对这些问题的研究。如今,基因功能注释领域中的自动功能预测(automatic function prediction,AFP)飞速发展,虽然还不足以解决上面提到的新蛋白质序列表征的问题,但是也已经提出一些类似于CASP 竞赛性质的比赛,如CAFA[97]、EFI[98]和COMBREX[99]等。相信在未来,会出现具有足够精度的人工智能算法能准确预测新蛋白质序列的功能,为人工智能辅助的蛋白质工程提供大量优质的数据。除此之外,随着微流控筛选、荧光激活的细胞分选、噬菌体辅助连续进化等超高通量筛选技术的突破与二代测序技术的成熟,二者联用产生的蛋白质深度突变扫描技术应运而生[100-102],应用它们来获得大量更全面、更均匀的实验数据是未来重要的发展方向之一。并且,近几年人工智能算法仍在飞速发展,迁移学习模型取得了一些进展,除了Frances H.Arnold 团队和George M.Church 团队所采用的自然语言算法模型外,自动编码器和变分自编码器神经网络算法也可以从输入的蛋白质序列中生成、提取深层的特征,从而基于序列就可以执行多种预测任务。例如Debora S.Marks 团队开发的DeepSequence 仅基于序列就可以预测突变带来的影响[103]。最后,人工智能算法的可解释性也是重要研究方向。相信在未来,能够清晰明了地解析预测模型内部原理。随着数据和人工智能算法的不断发展,性能更好的人工智能预测模型将会成为蛋白质工程的强大工具。
