亚胺还原酶在手性胺合成中的应用
2022-07-15杨璐瞿旭东
杨璐,瞿旭东
(上海交通大学生命科学技术学院,微生物代谢国家重点实验室,教育部代谢与发育科学国际合作联合实验室,上海 200240)
手性胺作为手性助剂以及关键中间体[1-4],在医药、精细化工、农业等领域具有重要的应用价值。据统计超过40%的手性药物中均含有手性胺结构单元[5],其中,具有环状结构的手性胺因同时具有胺的特性和杂环化合物的特殊活性,被作为中间体广泛应用于天然产物和手性药物的合成,例如新型喹诺酮类抗菌药物加雷沙星(Garenoxacin)以及用于治疗膀胱过度综合征的索非那新(Solifenacin)等(图1)。手性胺可以通过化学合成、酶催化、外消旋体拆分及组合催化等方法合成[6-10]。相较于传统化学合成方法,酶催化具有反应条件温和、催化效率高、立体选择性强、环境友好等优点,近年来被广泛应用于手性胺的合成中[11-13],在药物研发与生产领域展示出巨大的潜力。
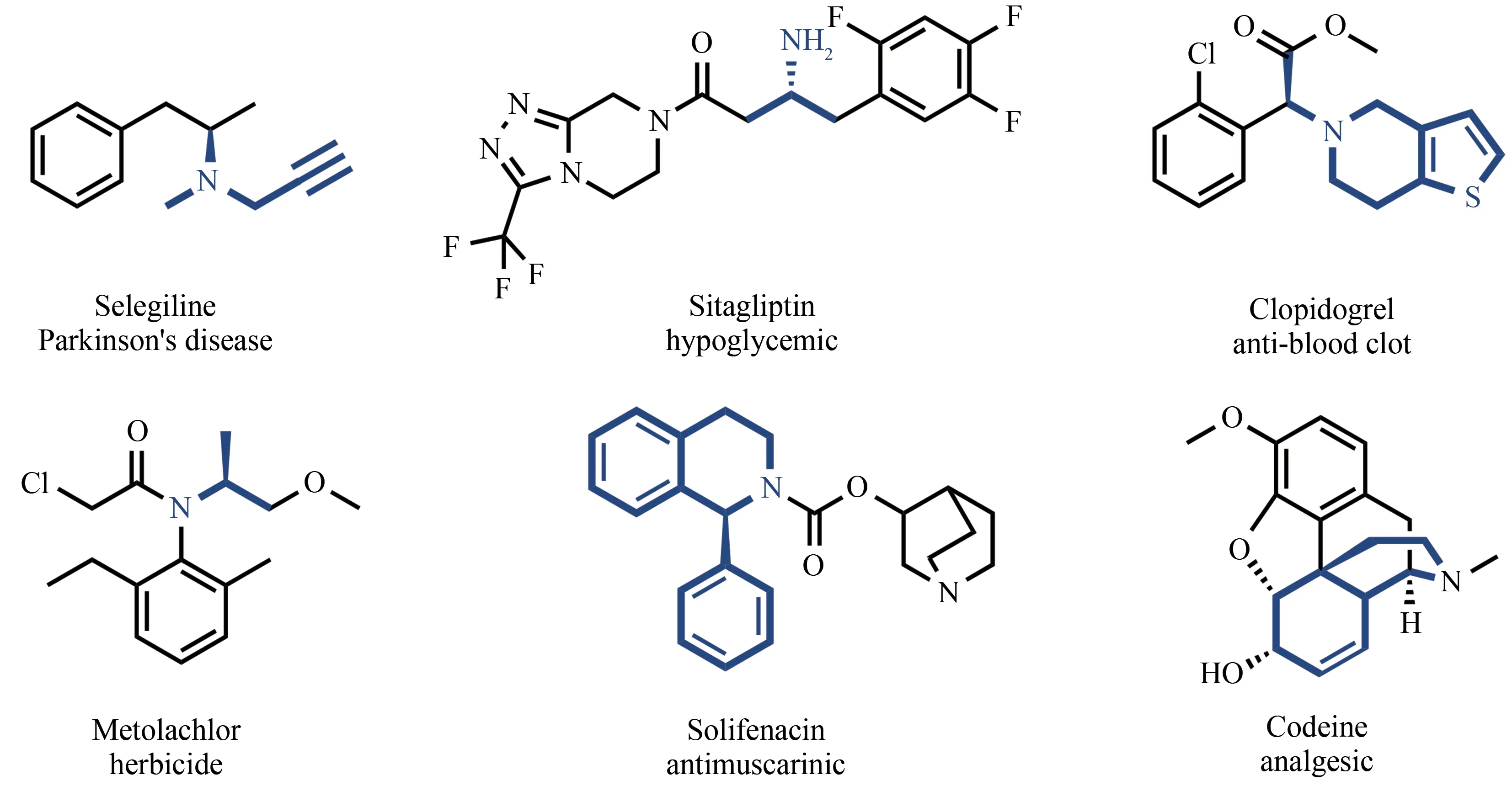
图1 含有手性胺结构的药物Fig.1 Pharmaceuticals containing chiral amine motifs
目前已报道的可用于手性胺合成的酶有脂肪酶[14-15]、转氨酶[16-19]、单胺氧化酶[20-21]、氨基脱氢酶[22]、亚胺还原酶[23-25](含还原胺化酶)和裂解酶[26-27]等。与转氨酶(只产生伯胺)、氨基脱氢酶(底物谱有限)等其他家族的酶相比,亚胺还原酶(IRED)具有极高的立体专一性和更广泛的底物选择性,不仅适用于合成手性伯胺,还能用于仲胺以及叔胺产物的合成[28]。此外,IRED 还被广泛用于还原环状亚胺合成手性含氮杂环,因此IRED的研究及应用备受学术界与工业界的关注。本文总结了IRED的发掘、开发以及在催化合成手性胺方面的研究进展,并对IRED的未来发展方向进行了展望。
1 亚胺还原酶的发现与催化机制
亚胺是指含有碳氮双键(C===== N 键)的有机化合物;在生物体内亚胺常出现在含氮化合物的代谢过程中,其经过还原酶的作用被还原成胺化合物用于合成代谢产物,例如叶酸[29][图2(a)]、铁载体[30-31][图2(b)]以及吗啡合成途径的关键中间体牛心果碱(Reticuline)等[32-33][图2(c)]。尽管对亚胺的还原过程早已熟知,但是由于还原酶的底物普适性较低,限制了这些酶对其他亚胺底物的应用拓展。同时亚胺在水中极不稳定,对于IRED 的筛选工作带来了巨大挑战,导致近10 年才有了较大突破。
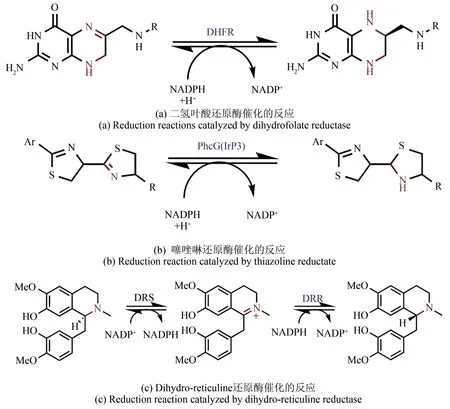
图2 天然代谢过程中的亚胺还原过程Fig.2 Imine reduction in the biosynthesis of natural products
1.1 亚胺还原酶的发现
IRED 是一类可将前手性亚胺不对称还原为相应胺化合物的NAD(P)H 依赖的氧化还原酶。1988年,Nardini 等[34-35]从猪肾和牛脑分离得到了能将环状酮亚胺催化生成手性环状氨基酸的酮亚胺还原酶,之后Hallenh等[36]又从人体中发现了类似的酮亚胺还原酶。随着研究的深入,在微生物体系中也发现了类似的酮亚胺还原酶:2004年Li等[37]对亚胺底物采用动态组合筛选方法,发现了厌氧菌Acetobacterium woodii具有亚胺还原酶活性,可将苄苯亚胺和丁苯亚胺还原为相应的胺。同年Muramatsu 等从Pseudomonas putidaATCC 12633 分离得到一个还原酶,可高效催化L-氨基酸酮酸的还原氨化生成R-甲基氨基酸[38];随后该课题组又从该菌株中发掘出编码苹果酸/L-乳酸脱氢酶基因dpkA,在大肠杆菌中异源表达并对其产物进行了表征,发现该酶既不具有苹果酸脱氢酶活性,也不具有乳酸脱氢酶活性,但可分别催化亚胺合成L-哌酸盐和L-脯氨酸[39-40]。2008 年Vaijayanthi 等[41]报道了一种高对映选择性的不对称还原芳基亚胺的生物催化方法, 利用近平滑假丝酵母Candida parapsilosisATCC 7330 催化产生R构型芳香仲胺。2010年Espinoza-Moraga等[42]筛选到能够不对称催化还原β-咔啉亚胺的贝氏酵母Saccharomyces bayanus,以45%~68%的收率和50%~97% ee 的立体选择性得到了一系列手性胺类产物。
2011 年Mitsukura 等[43-44]发现了几株来源于链霉菌的具有亚胺还原活性的氧化还原酶,能够催化不对称还原2-甲基吡咯啉生成2-甲基吡咯烷。其中来自Streptomycessp.GF3587 的R型亚胺还原酶(RIR)能高效催化2-甲基吡咯啉生成R构型产物,而来自Streptomycessp.GF3546 的S型亚胺还原酶(SIR)则可生成S构型产物。2-甲基吡咯啉结构简单,而且在水中稳定性高,这一研究迅速推动了亚胺还原酶在环状亚胺不对称催化的发展。随后Turner 等[45]将这两例亚胺还原酶在大肠杆菌中进行了异源表达,实现了多种环状亚胺的不对称还原,拓展了底物图谱,并报道了在亚胺不对称还原和生物催化级联反应的应用。RIR 的重组活性很高,且底物普适性也较好[46];SIR 虽然活性较低,但仍能高效催化五元环、六元环、七元环亚胺及多环二氢异喹啉的不对称还原[47]。至此,两个亚胺还原酶为后续亚胺还原酶的基因挖掘提供了重要参考,越来越多的亚胺还原酶被鉴定报道出来,大大拓展了亚胺还原酶家族[48-52]。
2012 年Meneely 等[30]从Yersinia enterocolitica分离得到了一株NADPH 依赖的噻唑啉还原酶(Irp3)并解析了其晶体结构。2013 年,Mirabal-Gallardo等[53]在Eisenia foetida上清液中实现了β-咔啉类亚胺的不对称还原,得到高立体选择性的R构型化合物。2016年Peng等[54]在研究聚酮合成过程中发现了一步不寻常的亚胺还原过程,该酶属于酰基辅酶A 脱氢酶家族,是一类新型的亚胺还原酶,为生物碱的生物合成提供了一条新的途径。同年Li等通过基因序列比对,构建并表达了15个亚胺还原酶,以3H-吲哚类化合物为模式底物进行筛选,成功找到性能较好的3个亚胺还原酶PlSIR、PlRIR和SnIR,并进行了酶改造,提升了催化活性及立体选择性[55];随后他们又鉴定了SnIR对于1-甲基-3,4-二氢异喹啉的催化效率较其他亚胺还原酶有25~1400倍提升[56]。2017年Zhu等[24]构建了含有88个亚胺还原酶的大型酶库,以1-芳基取代的二氢异喹啉化合物为底物进行筛选,成功挖掘了一组耐受大空间位阻二氢异喹啉化合物的亚胺还原酶,其中IR45、IR2能分别催化产生S和R构型化合物,ee值>99%。同年,Aleku 等[23]从米曲霉中发现了一个IRED同系物,显示出强大的还原胺化活性,将其命名为RedAm。这一发现使得羰基化合物和胺的分子间还原胺化成为可能,开辟了新的领域。2020 年Zhang等[57]通过对天然亚胺还原酶的研究,发现了能够识别2-芳基取代吡咯并且能催化得到两种构型产物的亚胺还原酶。利用具有R立体选择性的ScIR和S立体选择性的SvIR,合成了多个具有良好对应选择性的2-芳基取代的吡咯烷类手性中间体。
1.2 亚胺还原酶的结构及催化机理
由于IRED 具有巨大的应用前景,而理解其结构基础有助于酶工程的应用提高IRED 的性能。Rodríguez-Mata 等[58]解 析 了 来 源 于Streptomyces kanamyceticus的R型IRED Q1EQE0 的 晶 体 结 构(PDB:3ZHB)。IRED 由两条相同的单体通过相互作用组成二聚体,每个单体的N 端区域与NAD(P)H 结合形成罗斯曼折叠(Rossman-fold),并通过一个长螺旋连接到C 末端(图3);IRED 的活性口袋处于两个单体的N端结合部。

图3 R-IRED Q1EQE0的同源二聚体的结构Fig.3 Structure of the R-IRED Q1EQE0
结构上Q1EQE0 与一类β-羟基酸脱氢酶HIBDH(PDB:2CVZ)[59]高度类似。HIBDH 同样属于NAD(P)H 依赖的氧化还原酶,但催化的反应不同;其催化酮酸还原形成羟基酸。在催化过程中,HIBDH首先利用NAD(P)H的活性氢对酮酸的羰基进行亲核加成生成氧负离子,而后再由165位的赖氨酸提供质子将氧负离子转化成羟基并完成催化[图4(a)]。对比HIBDH 和Q1EQE0 的晶体结构,HIBDH 中165 位赖氨酸在Q1EQE0 中对应为187 位的天冬氨酸,并且该氨基酸在绝大多数的RIRED 中保守。 基于HIBDH 的催化机制,Rodriguez-Mata 等[58]推测了R-IRED 的催化机制:其利用187 位的天冬氨酸与亚胺的氮原子形成氢键,将亚胺底物固定住,而后再由NAD(P)H 的活性氢对亚胺进行亲核加成,生产氮负离子,再由周边的电正性氨基酸或水分子提供质子,最后将氮负离子转化成胺并完成催化[图4(b)]。
随着第1例IRED晶体结构被解析出来,陆续又报道了多个亚胺还原酶的晶体结构,如来自Streptomycessp.GF3546 的S-IRED(PDB:4OQY),来 自Streptomyces aurantiacus的S-IRED(PDB:4OQZ), 来 自Bacillus cereus的S-IRED(PDB:4D3F,4D3D),来自Nocardiopsis halophila的S-IRED(PDB:4D3S),来自Pseudomonas putidaKT2440 的RIRED (PDB:3L6D) 以 及 来 自Mycobacterium smegmatis的S-IRED(PDB:6SMT)[60-63]等。与R型IRED 对比,S型IRED 对应的催化位点为酪氨酸。为了验证该位点的作用,Rodríguez-Mata 等[58]对IRED 进行了一系列点突变实验。将R-IRED 中的187 位的天冬氨酸突变为不带电荷的丙氨酸和天冬酰胺后,酶失去了活性;类似的,将S-IRED 的酪氨酸突变为不带电荷的丙氨酸,酶也失去了催化活性,这些实验证实了这两个氨基酸位点对催化活性的重要作用。基于该结果,Rodriguez-Mata 等随后推测了S-IRED 的催化机制:其利用酪氨酸与亚胺形成氢键起到固定底物和极化亚胺键的作用,而后再由NAD(P)H 的活性氢对亚胺进行亲核加成,最后通过酪氨酸的酚羟基提供质子形成胺并完成催化过程[图4(c)]。
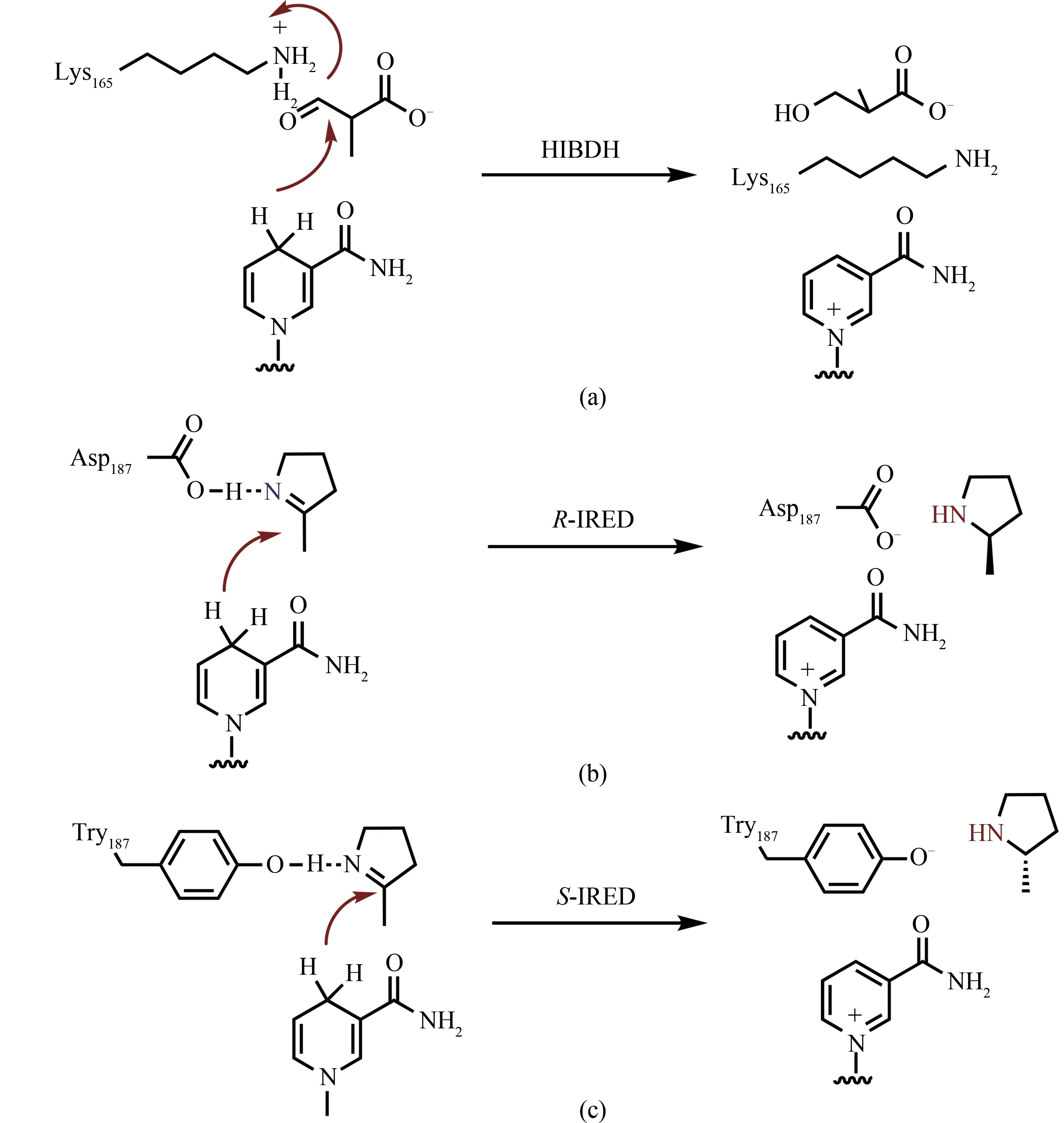
图4 HIBDH、R-IRED和S-IRED催化机理Fig.4 Catalytic mechanisms of HIBDH,R-IRED and S-IRED
除了催化机制外,Zhu 等[24]为了探究R-IRED和S-IRED结合位点的差异,以Q1EQE0为模板,模拟得到了可以形成S构型化合物的IR45和形成R构型化合物IR2的结构模型,并以1-芳基二氢异喹啉为底物进行了分子对接。结果表明,底物位于IR45的活性口袋中,被183位的天冬氨酸和NADPH所包围,符合之前推测的机理。同时底物的苯环指向191位的色氨酸和254位的甘氨酸形成的空腔,这种朝向使得底物分子位于NADPH的Si面,利于形成S构型化合物。而与IR2的对接结果表明底物的苯环指向右上的活性空腔,从而利于形成R构型化合物。该工作对IRED催化1-芳基二氢异喹啉的立体选择性提出了一种较为可靠的模型。
2 亚胺还原酶的改造
野生型的酶一般存在底物选择性窄、酶活性较差等局限,为了获得催化活性良好的酶,需要进行酶工程改造[64]。酶工程改造,主要包括酶分子的理性设计和非理性设计。其中理性设计包括定点突变、拼接和从头设计[65]。非理性设计是构建随机突变或重组文库,对该多样性文库的基因产物进行筛选,通过定向进化的方式获得编码改进功能产物的基因,同时用来继续下一轮进化,直至获得目的酶突变体。表1列出了一些较为典型的亚胺还原酶改造案例。
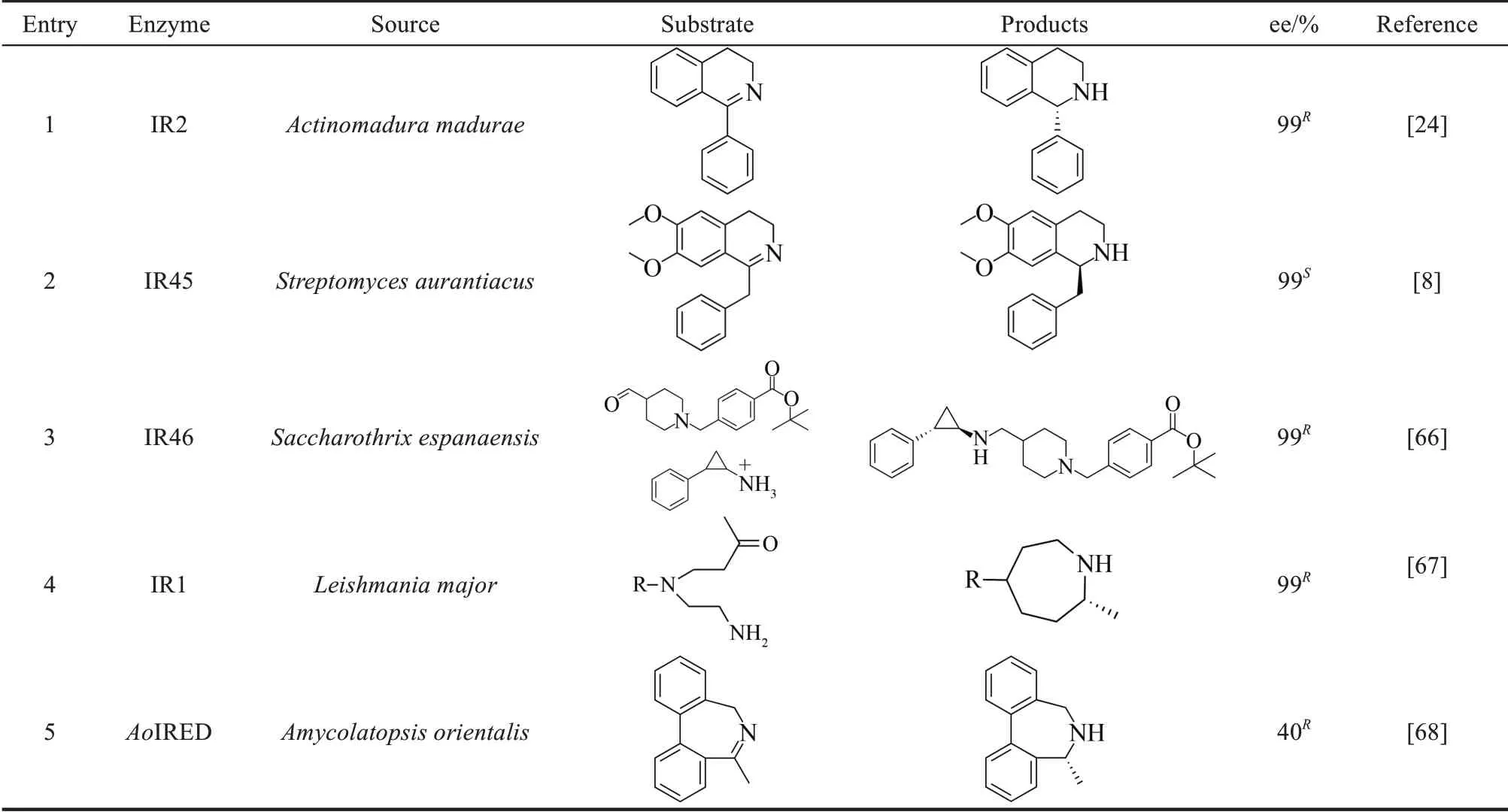
表1 亚胺还原酶改造实例Tab.1 Examples of imine reductase modifications
2.1 定向进化
定向进化是一种强大的酶工程方法,在近20 年的化学合成中取得了巨大的成功[69]。通过反复诱变和筛选,可以有效优化酶的性质,如对非原生底物的活性、底物选择性、产物选择性和稳定性,并巧妙地绕过对酶序列和结构、活性物质和反应机制不清楚的限制。但使用该方法筛选量大,一般需要与高通量筛选方法相结合[70]。
Hestericová 等[71]采用定向进化的方法,对一株人工设计的亚胺还原酶进行改造。采用无细胞提取液代替纯酶的催化方法,筛选了300 个突变体。在几轮进化中,找到了5个催化活性增强,对环状亚胺还原性较强的关键位点S112、N118、K121、S122 和L124Y,成功获得了2 个效率较好的多位点组合突变体,S112A-N118P-K121A-S122M和S112R-N118P-K121A-S122M-L124Y。该突变体相较于野生型酶,可得到较高立体选择性的R构型产物(95%ee)和S构型产物(86%ee)。
Schober 等[66]对1 株野生型IRED 进行了定向进化改造,通过3轮进化,使得突变体催化效率较野生型提高了38 000 倍,并且以84%收率得到赖氨酸特异性去甲基化酶-1 (LSD1) 抑制剂GSK2879552的关键中间体,光学纯度高达99.9%,发挥了酶在工业化生产带来的经济、绿色可持续、高产量的优势。
2.2 理性设计
对于结构和功能已知的酶进行改造,可以采用理性设计的方法来提升酶的活性[72]。一般而言,与底物结合和参与反应的关键氨基酸残基均在活性口袋附近,对这些关键氨基酸残基进行突变可以改变酶的性质。虽然远离活性中心的突变有时可提升酶的活性和热稳定性,但是对活性口袋周围的氨基酸突变影响明显高于远离活性中心位置[73]。肌氨酸是一种N-甲基化氨基酸,可用于合成抗精神病类药物。Mindt 等[74]从Pseudomonas putida分离得到IRED DpkA,并对底物结合位点进行定点突变,得到突变体DpkA-F117L,提升了乙醛酸的甲基化和乙基化的比活性(较野生型提升了1.18 倍),使该突变体在生产菌株中表达时可以更快地发酵生产肌氨酸,容积生产率可达0.16 g/(L·h)。天然来源的酶活性通常不高,底物选择性较窄,通常要经过改造来提升酶的活性及选择性[75]。提高立体选择性可以简化有机合成的下游提纯步骤。Aleku等[23]通过对特殊亚胺还原酶AspRedAm(又称还原胺化酶)晶体结构进行分析,找到了影响产物构型的关键氨基酸位点W210,单突变体W210A 实现了R构型向S构型的反转。Zhu 等[24]将野生型IR45与1-苯基-1,2-二氢异喹啉进行分子对接,发现W191 对于空间位阻大小和维持酶活性至关重要,对该位点进行饱和突变后,找到了一个优势突变株W191F,酶活力较野生型提高了1.5倍。
四氢异喹啉生物碱是一类庞大的手性含N 杂环化合物,是很多药物和天然产物的分子骨架。Yang 等[8]通过IR45 与二氢异喹啉底物的分子对接模拟,找到了两个关键的氨基酸残基位点F190 和W191,并对其进行了饱和突变,成功筛选到两个催化活性优于野生型的突变体。其中IR45-F190LW191F 对于两个空间位阻最大的化合物实现了100% 转化且ee 值>99%,而野生型没有活性。IR45-F190M-W191F 对于底物的催化活力也有1.8~6.8倍提升,转化率可达100%,ee值>99%。
手性七元氮杂环在天然产物和药物具有广泛的应用,其中手性1,4-二氮杂烷是重要的结构单元,具有多种生物学特性和药用价值,例如治疗青光眼和高压眼症的Ripasudil[76]。Xu 等[67]通过酶库筛选,获得了一系列具有较好催化活性和立体选择性的IRED。进一步对其中的一个酶IR1 进行了改造和迭代组合突变,得到双突变体Y194FD232H,其催化效率是野生型酶的61 倍,底物投料浓度可达100 mmol/L,具有工业化前景。该工作为通过IRED 催化相应氨基酮的分子内还原胺化为手性1,4-二氮杂烷提供了一种有效的方法。
尽管一些IRED 偏好NADH[77],大部分IRED仍依赖于NADPH。由于NADPH 的价格远高于NADH,因此IRED 对于NADPH 的偏好性,限制了其在生物催化中的应用。因此辅因子偏好性的改造,对于IRED 在工业应用拓展方面有着重要作用。Gand 等[78]对对从链霉菌GF3587 分离得到的IR-Sgf3587 进行了合理的设计,从15 个突变体中筛选到1 个单突变体K40A,使得酶对NADH 依赖度增加了3 倍,并且利用该突变体实现了2-甲基吡咯啉的制备级转化,转化率可达88%。为了改变NADPH 到NADH 的 特异 性,Borlinghaus 等[79]以 来 自Streptomyces kanamyceticus的R-IRED(PDB:3ZHB,同源性42%)为模板,应用了“辅因子特异性反向结构分析和库设计”(CSRSALAD)诱变策略,得到了野生型的R-IRED模型[图5(a)],并对R-IRED 的6 个氨基酸位点进行改造[图5(b)],将NADH 的偏好性提高了900 倍。在此基础上将37 位赖氨酸(K37)进一步突变为丙氨酸[图5(c)],最终将NADH 的选择性提高了2900倍。
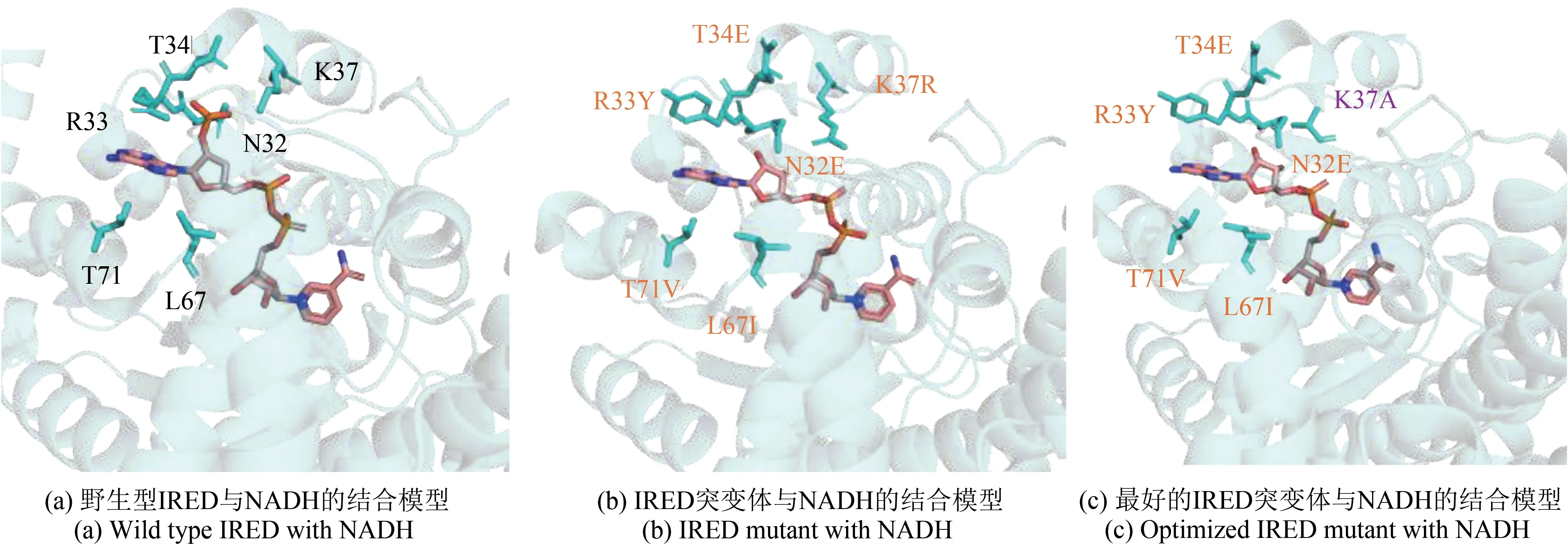
图5 R-IRED及突变体与NADH的对接模型Fig.5 Docking models of R-IRED and its mutants with NADH
3 亚胺还原酶的应用拓展
在工业生物技术中的生物催化具有化学催化无可比拟的优势。例如酶的专一性强,具有高效的底物选择性,包括立体选择性、区域选择性及化学选择性;酶法反应条件温和,反应能耗低,较化学方法可控度高;酶法对于环境友好,减少了相应的上保护、脱保护等步骤,可降低成本,减少污染物排放。
IRED 的研究虽然起步发展较晚,但随着测序技术的发展,每年数据库收录的基因序列越来越多,大量未知功能的IRED 被挖掘出来,在合成应用方面取得了不少成效。
3.1 多酶串联的合成应用
IRED 与其他功能酶串联,可以实现更广泛的功能,得到更多复杂化合物的生物合成。Heath等[80]开发了一种胺氧化酶-亚胺还原酶体系。通过将R-6-羟基-D-尼古丁氧化酶(6-HDNO)与亚胺还原酶R-IRED 偶联,实现了一系列哌啶衍生物和吡咯烷的去外消旋化,产生高收率和对映体过剩值的S构型胺化合物。France 等[68,81]开发了一种IRED 和ω-转 氨 酶(ω-TA)合 成dibenz[c,e]azepine 骨架的方法,并对亚胺还原酶进行改造,提升了酶的活性和立体选择性。又开发了羧酸还原酶(CAR)、ω-转氨酶(ω-TA)和IRED 的生物催化级联反应,获得了手性单取代和双取代的哌啶和吡咯烷。利用这个策略,可以从酮酸或酮醛开始,一锅(one-pot)合成各种取代的哌啶或吡咯烷,并且每种酶在催化过程中均表现出高度化学选择性、区域选择性和立体选择性(图6)。Hepworth 等[82]将该级联方法应用于全细胞催化中,以较高转化率(>93%)和对映体过量值(>93%)得到一系列哌啶衍生物。Montgomery 等[83]将还原胺化酶(AspRedAm)与醇脱氢酶(ADH)偶联,实现了外消旋的醇到手性胺的过程。Ford等[84]利用半乳糖氧化酶(GOase)和IRED 的级联作用,合成了苄氧基羰基保护的氨基氮杂烷和氨基哌啶,一锅法避免了潜在的外消旋化合物形成不稳定中间体,最终得到54%收率的高对映体纯度产物。
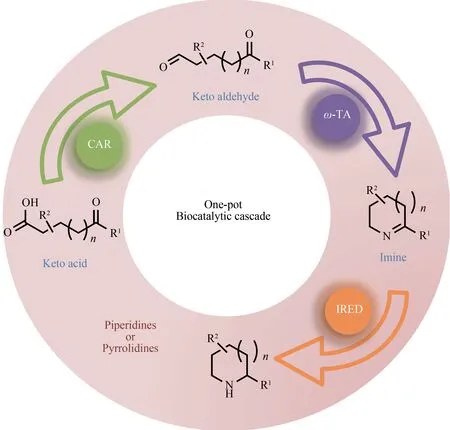
图6 一锅法多酶串联合成哌啶和吡咯烷Fig.6 One-pot cascade synthesis of piperidines and pyrrolidines
3.2 化学-酶串联的合成应用
化学与酶相结合是一种更简单、高效、经济的方法,既具有化学方法合成原料经济易得、合成工艺简捷高效的特点,又兼具酶法催化效率高、立体选择性好的优势,使其在高附加值手性化合物的合成方面得到广泛应用。
Cosgrove 等[85]开 发 了 一 种IRED 与 钯 发 生Buchwald-Hartwig 交叉偶联反应生产N-芳胺的方法,最终转化率可达90%,并且IRED 催化步骤建立的手性中心不受后面交叉偶联反应的影响。Yang 等[8]通过一步简单的化学合成,高效地得到一系列二氢异喹啉底物,再与IRED 和甲基转移酶串联,一锅法可完全转化得到N-甲基化的四氢异喹啉生物碱(图7)。

图7 化学-酶法高效合成四氢异喹啉生物碱Fig.7 High efficient synthesis of tetrahydroisoquinoline alkaloids by the chemo-enzymatic method
3.3 其他应用
发展更快速、更简单的筛选蛋白质序列空间的方法,来鉴定用于不对称合成的IRED,是目前面临的挑战和限速步骤。IRED 不对称催化制备手性胺的策略愈发诱人,为许多高价值化合物的生物合成提供了一条有效的途径。Marshall 等[86]发现了300 余种新的IRED,建立了包含384 种序列多样的用于筛选的IRED库,对36个氨基底物进行高通量筛选,通过动态动力学拆分实现了高收率和高立体选择性的N-取代β-氨基酯衍生物的制备级合成。也是迄今为止报道的最大的关于IRED 的鉴定、克隆、生产和合成应用方法。
4 总结与展望
手性胺是非常重要的结构单元,广泛存在于天然产物、药物、表面活性剂和农用化学品中。由于手性化合物的不同对映体对生物体的生理活性不同,分离和制备纯净的对映体一直以来都是研究热点和难点。通过外消旋体拆分法制备手性胺化合物,是目前工业上应用最为广泛的方法,选择合适的拆分试剂进行多次拆分纯化,进而得到所需构型手性胺。但是该方法理论上仅有50%的收率,会造成原料的浪费及成本的增加。亚胺的化学不对称还原通常采用金属催化或小分子催化方法[87-91],但金属催化剂不仅价格昂贵,对环境也会造成严重污染。随着酶催化方法的脱颖而出,IRED被越来越广泛地应用于手性胺的合成。
IRED 发展历史较短,但随着近10 年基因测序手段及酶工程改造技术的飞速发展,越来越多的功能性IRED 晶体结构被解析报道出来。IRED 在环状亚胺的不对称催化合成方面取得了骄人成绩,效果明显优于拆分和去消旋化,吸引了众多研究者的目光。同时胺基供体选用有机胺,大大增加了产物的宽泛性,逐渐成为合成手性胺的重要的途径之一。为了获得更具价值的化合物,IRED 可与其他酶偶联,发展多酶级联策略,这无疑赋予了IRED 更大的发展空间。将化学与酶法结合制备手性胺,可以极大丰富前体化合物骨架,提前预装目的官能团,有效避免了天然合成路径中某些活性差、表达量低的酶参与,将化学合成的优势与酶催化的特点相结合。
虽然IRED 在不对称催化合成手性胺方面得以开发和利用,但仍面临着不小的挑战。目前已知的IRED 晶体对于理解其立体选择性仍缺乏有力解释。此外,已报道的IRED 普遍存在底物选择性不够宽泛、稳定性不高、酶活力不足等问题,需要发展高通量筛选、酶工程改造等技术,不断对酶进行优化,扩大底物图谱,提高立体选择性。目前已知的IRED 大多是NADPH 依赖的,仅有极少数是NADH 依赖,虽然有一些方法可以改变辅因子的选择性,但是这些策略的通用性还需要验证。此外,IRED 对于非环状亚胺的活性远小于环状亚胺,因此提高IRED 在非环状亚胺反应中的应用,也是未来重要的研究方向。其次,复杂化合物体外酶催化全合成途径中,与IRED 串联反应的其他功能酶,可能面临酶不兼容、反应条件不合适、中间产物或辅因子抑制酶活性等问题,很大程度上影响多酶串联的级联反应效率。相信随着合成生物学、蛋白质改造和酶工程的不断发展,IRED的应用发展将愈加成熟,成为复杂化合物合成的重要手段之一。
