国产某品牌谷胱甘肽还原酶试剂盒多中心横向联合性能评价
2022-07-15朱宇清王海滨王连明冯品宁郭书忍彭亦冰程黎明传良敏0王华梁
朱宇清,王海滨,王连明,赵 芳,冯品宁,张 鹏,郭书忍,彭亦冰,程黎明,传良敏0,王华梁
(1.上海市临床检验中心临床生化免疫学研究室,上海 200126;2.中国人民解放军总医院检验医学中心,北京 100039;3.哈尔滨医科大学附属第一医院检验科,黑龙江 哈尔滨 150001;4.中国医学科学院北京协和医院检验科,北京 100730;5.中山大学附属第一医院检验科,广东 广州 510080;6.南方医科大学南方医院检验科,广东 广州 510515;7.郑州大学第一附属医院检验科,河南 郑州 450052;8.上海交通大学医学院附属瑞金医院检验科,上海 200025;9.华中科技大学同济医学院附属同济医院检验科,湖北 武汉 430030;10.四川省人民医院检验科,四川 成都 610072)
谷胱甘肽还原酶(glutathione reductase,GR)是人体氧化还原体系中最重要的酶之一,是维持细胞中还原型谷胱甘肽含量的主要黄素酶。在还原型烟酰胺腺嘌呤二核苷酸磷酸的参与下,氧化型谷胱甘肽会转化为还原型谷胱甘肽,后者在防止血红蛋白的氧化分解、维持巯基蛋白的活性、保证巯基蛋白的还原性及细胞的完整性等方面发挥重要作用[1]。GR可用于葡萄糖-6-磷酸脱氢酶缺乏症、慢性肾功能衰竭、糖尿病、心脑血管疾病等多种疾病的辅助诊断[2-5],也可用于肿瘤化疗药物的耐药性观察[6]。近年来的研究结果表明,GR水平升高与多种肝脏疾病相关,可作为临床诊断、评估肝损伤的辅助指标,对于肝脏损伤具有较好的预警作用[7]。
目前,GR活性检测已被广泛应用于临床,检测方法以紫外酶法为主,以氧化型谷胱甘肽为底物,在GR的催化作用下生成还原型谷胱甘肽,同时将还原型烟酰胺腺嘌呤二核苷酸磷酸氧化成氧化型烟酰胺腺嘌呤二核苷酸磷酸,通过检测还原型烟酰胺腺嘌呤二核苷酸磷酸的下降速率来测定GR活性。
本研究在全国范围内采用多中心横向联合评价的方法,对江西乐成公司GR检测试剂盒进行了性能评价。所有参与评价的实验室必须在相同的时间,按照统一的评价方案,使用相同批号试剂,对同一批标本进行测试和评估,最终汇总所有实验室的检测结果进行横向比较和综合评价,从而实现了对该品牌GR检测试剂的检测性能进行全方位、多维度的评价。
1 材料和方法
1.1 研究对象
选取我国不同地区9家三级甲等综合性医院临床实验室[中国人民解放军总医院(301)检验医学中心、哈尔滨医科大学附属第一医院检验科、中国医学科学院北京协和医院检验科、中山大学附属第一医院检验科、南方医科大学南方医院检验科、郑州大学第一附属医院检验科、上海交通大学医学院附属瑞金医院检验科、华中科技大学同济医学院附属同济医院检验科、四川省人民医院检验科],组成多中心联合评价小组,同步进行该品牌谷胱甘肽还原酶(GR)试剂盒检测性能评价。
1.2 仪器和试剂
9家临床实验室使用的全自动生化分析仪包括cobas c701 全自动生化分析仪(瑞士罗氏公司)3台、ARCHITECT c16000全自动生化分析仪(美国雅培公司)2台、AU5800全自动生化分析仪(美国贝克曼库尔特公司)4台。江西乐成公司谷胱甘肽还原酶(紫外酶法)检测试剂盒(批号20190426、20190118、20190308)、配套校准品(批号20190320、20190413)和质控品(批号20190320、20190413),英国朗道公司谷胱甘肽还原酶(紫外酶法)检测试剂盒及配套校准品(批号450780)。
1.3 方法
所有检测标本均为无溶血、黄疸、脂血的临床剩余血清。多中心联合评价小组按照统一的评价方案,使用各自实验室生化分析仪、江西乐成公司GR试剂和校准品组成的被评价检测系统进行实验。所使用的江西乐成公司GR试剂、校准品、质控品和英国朗道公司GR试剂均为相同批号。用于线性范围、最大稀释倍数和比对试验的血清样品均为相同的分割样品。实验时间严格控制在2019年7月8—12日,5 d内全部完成。
1.3.1 重复性 参照《CNAS-GL037 临床化学定量检验程序性能验证指南》[8]的建议,检测低(L1)、中(L2)、高(L3)3个浓度非定值质控品,在1批内分别连续检测20次,计算检测结果的均值()、标准差(s)、变异系数(coefficient of variation,CV)。重复性(CV)小于室间质量评价(external quality assessment,EQA)标准的1/4(5%),则验证通过。分别计算3个浓度质控品所有实验室重复性(CV)的。
1.3.2 室内精密度 参照《WS/T 492—2016临床检验定量测定项目精密度与正确度性能验证》[9]建议的精密度验证方案,用同一批试剂检测低(L1)、中(L2)、高(L3)3个浓度非定值质控品,每天检测3次,连续测5 d,计算批内标准差(sr)、批间标准差(sb),实验室内标准差(s)和实验室内CV。实验室内CV小于EQA标准的1/3(6.67%),则验证通过。分别计算3个浓度质控品所有实验室内CV的。
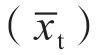
1.3.4 线性范围 取已知浓度的高(H)、低(L)值样品各1份,按5L、1H+4L、2H+3L、3H+2L、4H+1L、5H配制成6个浓度系列样品,分别重复检测3次。参照《WS/T 408—2012 临床化学设备线性评价指南》[11]建议的方案进行统计,剔除离群值后,将检测结果进行一次、二次和三次多项式回归分析,对非线性系数进行t检验,判断非线性系数与“0”是否有显著性差异。如果二次和三次多项式的非线性系数与“0”无显著性差异,则数据组被认为具有线性,此时可对数据组进行精密度检验,精密度符合线性判断要求时,可确定数据组具有统计学线性或一阶线性;如果非线性系数与“0”具有显著性差异,则进行非线性程度判断和精密度检验。计算最优拟合曲线与直线的平均差异值(average deviation from linearity,ADL),将ADL与临界值[11]进行比较,ADL小于临界值,则判定为临床可接受的非线性,即二阶线性;反之则判定为非线性。
1.3.5 最大稀释倍数 使用高值血清样品,按照厂家推荐的样品最大稀释倍数进行稀释,本研究为5倍稀释。稀释后,在1批内重复检测3次,计算,并以×稀释倍数后的值作为实际值,计算回收率,计算公式为:回收率=实际值/理论值×100%,以(100±10)%为可接受范围[12]。
1.3.6 偏移评估 参照文献[9],使用患者样品的正确度验证方案,以各实验室的生化分析仪、英国朗道公司GR试剂和校准品组成对照检测系统,进行比对试验。分别使用被评价检测系统和对照检测系统检测40份血清样品。样品浓度按试剂盒检测线性范围平均分布。计算被评价检测系统与对照检测系统之间配对结果的偏移。将数据进行配对t检验,确定不同系统间的平均偏移以及偏移的s。若偏移小于EQA标准的1/2(10%),则验证通过;否则,计算偏移验证值,若偏移在偏移验证值范围内,则验证通过。
1.3.7 试剂间比较 分别使用被评价检测系统和对照检测系统检测40份血清样品,将所有实验室的检测结果按照试剂分成国产试剂组和进口试剂组。分别比较国产试剂组和进口试剂组组内9家实验室检测结果的一致性,使用SPSS 25.0软件分别对9家实验室的检测结果进行Kendall相关性检验,Kendall协调系数为0.8~1.0表示极强相关,0.6~0.8表示强相关,0.4~0.6表示中等程度相关,0.2~0.4表示弱相关,0.0~0.2表示极弱相关或无相关。分别计算40份血清样品国产试剂组和进口试剂组的组内、s和CV,对组内和CV进行配对t检验,并进行相关分析。
1.3.8 仪器间比较 将40份血清样品检测结果按照试剂分为进口试剂组和国产试剂组,然后分别在2个试剂组内按照仪器品牌分为A、B、C 3个组。计算各仪器组内40份血清样品检测结果的、实验室间CV,对不同仪器组检测结果的进行两两配对t检验,比较相同试剂在不同仪器平台的检测结果之间的差异。
2 结果
2.1 重复性评价结果
各实验室检测低(L1)、中(L2)、高(L3)3个水平质控品的重复性(CV)均小于EQA标准的1/4(5%),9家实验室3个浓度质控品的平均CV分别为2.83%、2.42%、1.28%。见表1。
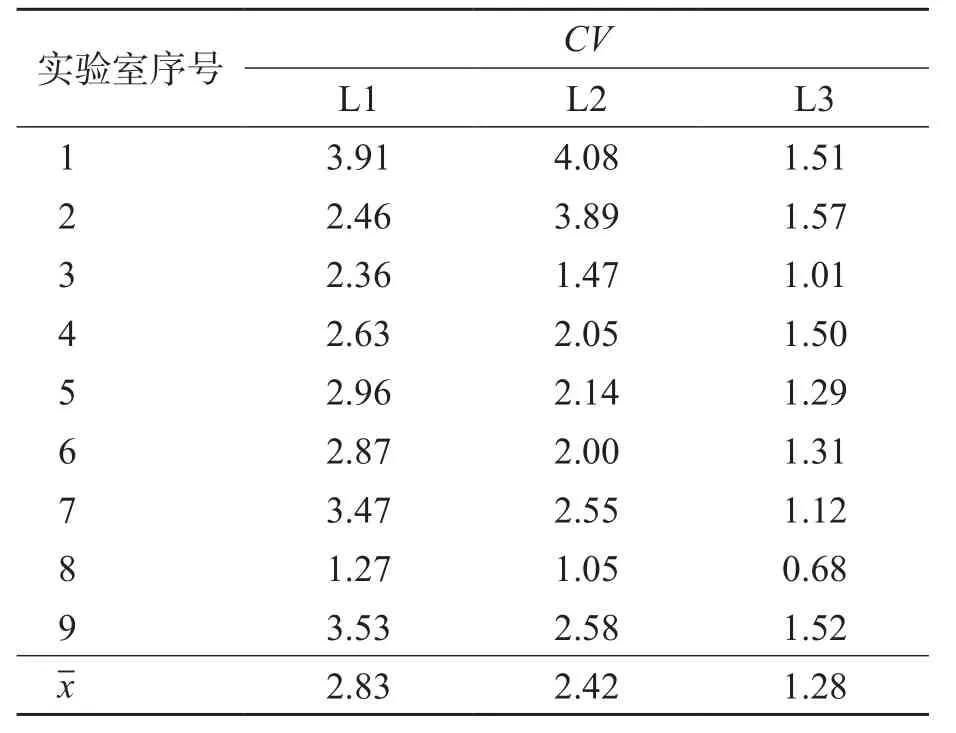
表1 重复性评价结果 %
2.2 室内精密度评价结果
各实验室检测低(L1)、中(L2)、高(L3)3个水平质控品的实验室内CV均小于EQA标准的1/3(6.67%),9家实验室3个浓度质控品的平均CV分别为3.15%、3.44%、2.07%。见表2。
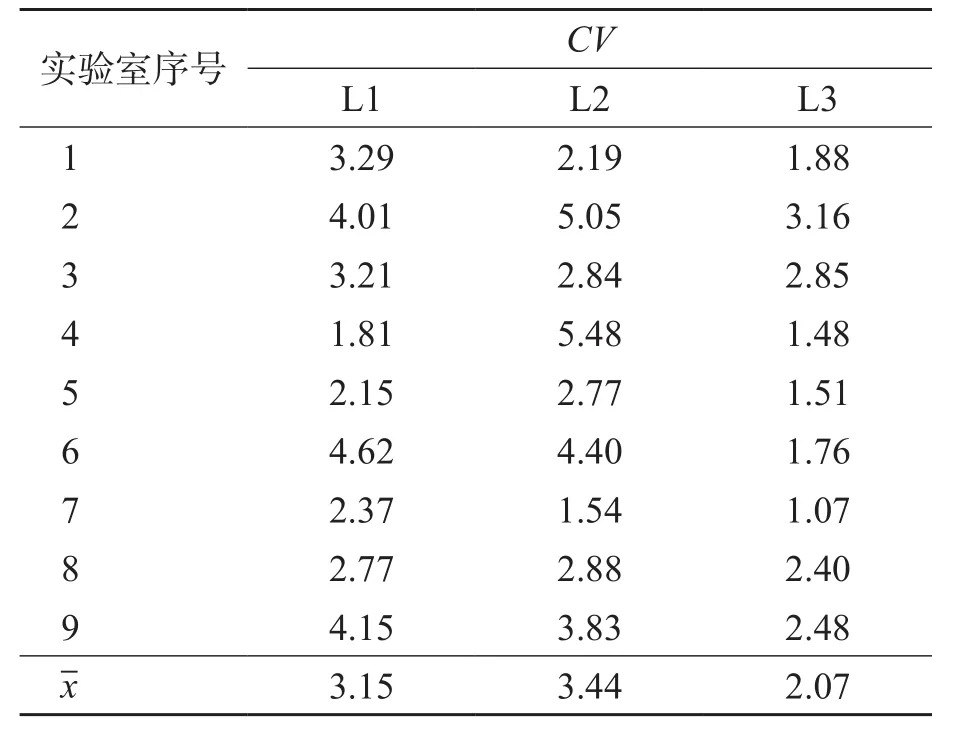
表2 室内精密度评价结果 %
2.3 试剂批间差
6号实验室低浓度(L1)样品相对偏差为11.6%,9号实验室中浓度(L2)样品的相对偏差为10.1%,其余实验室各浓度水平相对偏差均小于厂家声明(8%)。将所有实验室检测数据汇总后计算得到L1、L2、L3质控品的总体批间差分别为2.8%、0.5%、0.6%。见表3。

表3 试剂批间差评价结果 %
2.4 线性范围
在厂家声明的线性范围(10~320 U/L)内,3号、8号实验室最优拟合方程为二阶方程,7号实验室最优拟合方程为三阶方程,线性偏移均小于EQA标准的1/4(5%);其余实验室最优拟合方程均为一阶方程。所有实验室的相关系数(r)为0.997~1.000。见表4。
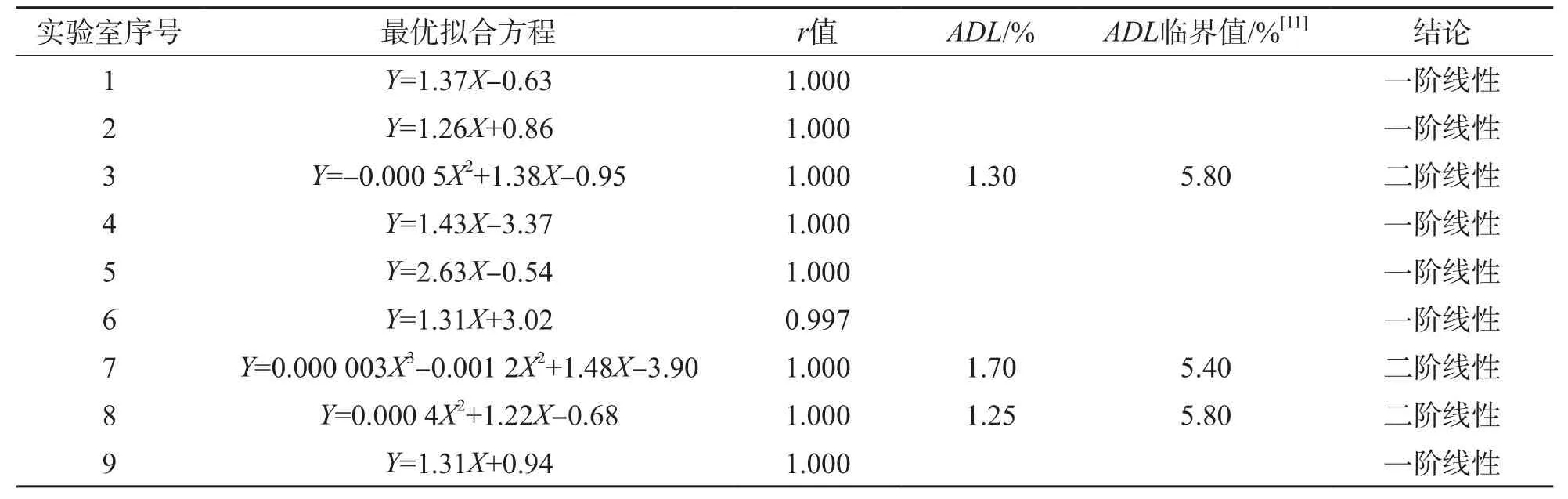
表4 线性范围验证结果
2.5 最大稀释倍数
将高值样品进行5倍稀释后进行检测,9家实验室的回收率分别为101.1%、100.9%、92.6%、106.3%、93.4%、103.4%、98.5%、97.4%、94.6%,平均回收率为98.7%,结果均在允许范围内。
2.6 偏移评估
9家实验室40份样品比对结果显示,偏移均小于EQA标准的1/2(10%),r值为0.997~1.000。见表5。
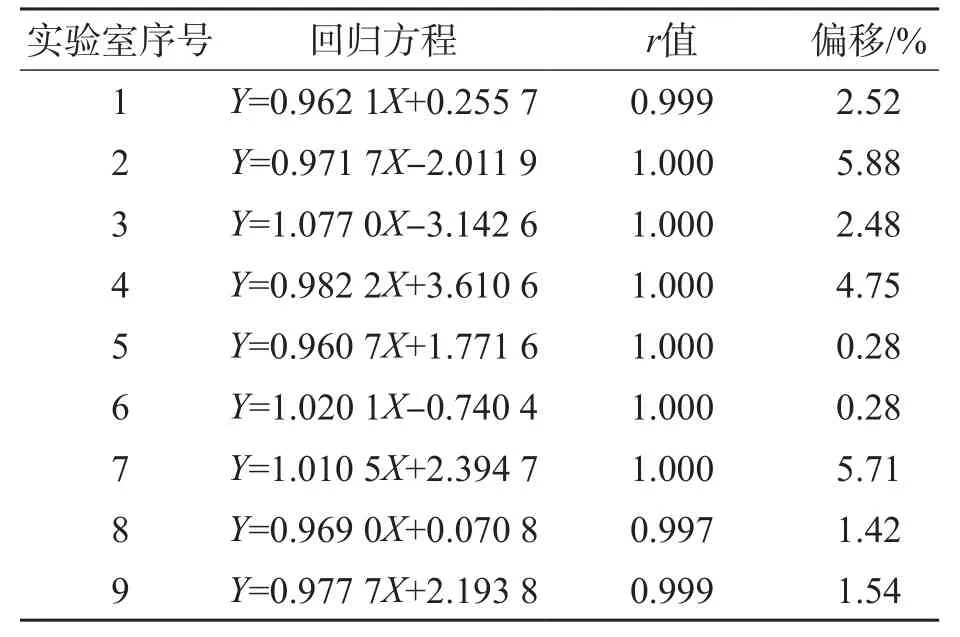
表5 偏移评估结果
2.7 试剂间比较结果
国产试剂组9家实验室检测结果呈中等程度相关,Kendall协同系数为0.471(P<0.01);进口试剂组9家实验室检测结果呈较强相关,Kendall协同系数为0.633(P<0.01)。40份血清样品国产试剂组与进口试剂组的组内CV差异无统计学意义(P>0.05);国产试剂组平均CV为6.48%,进口试剂组平均CV为6.68%。40份血清样品国产试剂组与进口试剂组的组内差异无统计学意义(P>0.05);以英国朗道公司GR试剂检测结果为X,以江西乐成公司GR试剂检测结果为Y,进行相关性分析,结果显示,2种试剂相关性良好(r=1.000,P<0.01)。见图1。

图1 2种GR试剂检测结果相关性
2.8 仪器间比较结果
国产试剂A仪器组与B、C仪器组检测结果差异均无统计学意义(P>0.05),B仪器组与C仪器组检测结果差异有统计学意义(P<0.05)。进口试剂组A仪器与B仪器组检测结果差异无统计学意义(P>0.05),C仪器组与A、B仪器组检测结果差异均有统计学意义(P<0.05)。见表6。
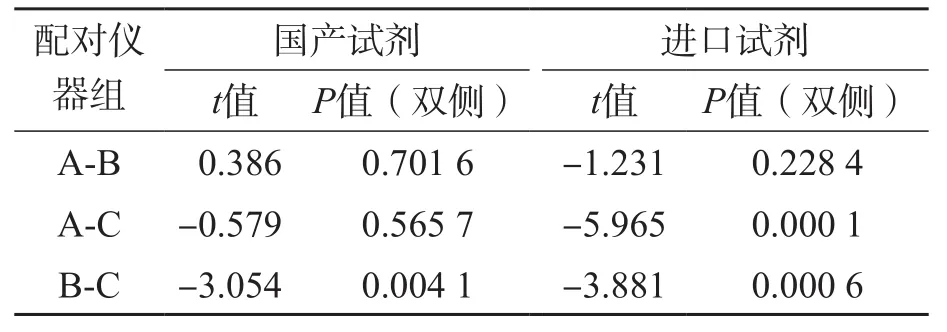
表6 仪器间比较
3 讨论
近年来,在国家层面相关科技计划的大力支持下,我国自主体外诊断领域已具备较好的产品研发基础,试剂创新与研制链已基本形成。传染病、肿瘤、心脑血管疾病、优生优育、代谢性疾病、内分泌疾病等系列国产诊断试剂已在临床广泛应用,部分试剂检测性能已达到或接近国际同等水平[13]。本研究在全国范围内联合多家三级甲等综合性医院实验室对国产试剂进行多中心横向联合性能评价,根据《CNAS-CL02-A003医学实验室质量和能力认可准则在临床化学检验领域的应用说明》[14],并参考相关卫生行业标准对江西乐成公司GR检测试剂的重复性、室内精密度、试剂批间差、线性范围、最大稀释倍数等性能进行了验证。新版《CNAS-CL02-A001 医学实验室质量和能力认可准则的应用要求》已于2021年05月25日发布,2021年11月25日开始实施,本研究开始进行此项评估时,新版要求还未发布,故本研究评价方案参照的是旧版本的文件[14],并且关于重复性精密度低于EQA标准的1/4,室内精密度低于EQA标准的1/3的要求,只在当时评估方案所采用的旧版本文件中才有要求,新版文件已无此要求。
本研究参照文献[8-9]要求,采用同一批试剂每天检测低(L1)、中(L2)、高(L3)3个浓度非定值质控品,验证重复性(在1批内分别连续检测20次)和室内精密度(每天检测3次,连续测5 d),结果显示,9家实验室重复性(CV)均小于EQA标准的1/4(5%),室内精密度(CV)均小于EQA标准的1/3(6.67%)。
试剂批间差评价结果显示,有1家实验室中浓度(L2)样品批间差超出了厂家声明。经过该实验室分析发现,用于批间差检测的主、副试剂开瓶日期不同步,主试剂于2019年7月8日开瓶,副试剂于2019年7月17日开瓶;另外,主、副试剂并未在同一时间段内进行定标,校准品7月8日复溶后用于主试剂定标,已复溶的校准品在冰箱内冷藏保存9 d后用于副试剂定标。按照厂家说明,校准品复溶后在2~8℃条件下稳定24 h。因此,该实验室试剂批间差结果不符合可能是不同批号试剂开瓶、定标不同步,以及副试剂定标所用校准品已过开瓶有效期所致。另有1家实验室的低浓度(L1)样品批间差超出厂家声明的允许范围。综合统计所有实验室的检测结果,3个浓度(L1、L2、L3)样品试剂总体的批间差均符合要求,试剂批间差性能基本满足要求,但仍有待进一步提高。
本研究中,有3家实验室线性范围评价验证结果的最适拟合多项式为二次多项式或三次多项式,统计学上为非线性,但是其ADL均小于临界值,故验证结果为临床可接受的非线性,即二阶线性。目前,GR项目既无参考测量程序,也无国际公认的有证参考物质,因此在本次联合评价中,我们参考了目前实验室普遍采用的卫生行业标准文件——《WS/T 492—2016 临床检验定量测定项目精密度与正确度性能验证》[9](此文件相当于CLSI EP15-A2文件,最新版的CLSI EP15-A3目前还没有相对应的行业标准),使用患者样品进行偏移评估的方案。选用英国朗道公司GR试剂作为参比试剂进行比对。所有实验室国产试剂和进口试剂检测结果相关性良好,偏移均在允许范围内,说明国产试剂与进口试剂具有可比性。
根据《CNAS-CL02 医学实验室质量和能力认可准则》[15]的要求,在常规应用前,应由实验室对检验程序进行独立验证,证实检验程序的性能指标。本研究中,9家实验室的验证结果显示,所评价的国产GR检测试剂盒的各项检测性能指标均达到了预期。多中心横向联合评价不仅可反映检测系统在单个实验室内的性能,还可以横向比较检测试剂测定结果在不同实验室之间的差异。本研究规定9家实验室在相同时间按照相同方案进行实验,使用的试剂、校准品、质控品均为同一批号,血清样品相同,即将每份样品平均分装成9份后分发到各实验室,因此9家实验室的检测结果具有一定的横向可比性。本研究将所有实验室的检测结果按照试剂品牌分组进行统计,结果显示国产试剂组和进口试剂组组内9家实验室的检测结果均具有显著的相关性,说明不同试剂组组内检测结果一致性较好;国产试剂和进口试剂的组内CV差异无统计学意义,说明国产试剂组实验室间的精密度与进口试剂组一致;国产试剂检测结果与进口试剂检测结果无明显差异,且结果具有显著相关性,说明国产试剂检测结果与进口试剂检测结果具有良好的一致性;仪器间比较结果显示,无论是国产试剂,还是进口试剂,在不同仪器平台的检测结果均存在一定差异,说明不同仪器平台之间存在一定的系统误差,提示厂家应根据不同仪器平台调整校准品赋值,以提高检测结果的一致性。
多中心横向联合评价作为一种全新的理念和评价方式,在不同地域、不同设备、不同环境、不同操作人员的条件下,使用同一样品在同一时间节点进行性能验证,是一种全新的尝试与探索。于临床实验室而言,参与多中心横向联合评价可以客观、全面地了解产品的综合性能,多中心横向联合评价的数据也有助于实验室对体外诊断试剂进行更加客观的了解;于患者而言,使用多中心横向联合评价通过的体外诊断试剂,在不同地域、不同医院、不同检测仪器条件下均能获得可比较的检测结果;于体外诊断研发企业而言,多中心横向联合评价有助于其了解试剂在不同检测系统上的差异,帮助其完善溯源体系,在不同检测系统上对校准品的赋值进行调整;于国家而言,多中心横向联合评价有益于推动我国民族企业体外诊断产品的品质提升,让国产体外诊断产品在市场上更具竞争力,惠及民族企业、医院与广大患者。同时,多中心横向联合评价也可为实现更大区域内的检验结果互认提供真实世界的数据,为促进这一政策的有效落地提供更切合实际的参考。
(本研究为多中心研究,所有作者对本研究具有同等贡献,作者排名按数据上报时间排序。)
