数据库分析和验证HMMR为乳腺癌的主要生物标志物
2022-07-13刘馨璥范苗苗毕佳欣
刘馨璥,范苗苗,赵 奇,毕佳欣,宋 洁
(牡丹江医学院基础医学院生物学教研室,黑龙江 牡丹江 157011)
乳腺癌是起源于乳腺导管或小叶的恶性肿瘤,在全球女性中发病率最高[1]。影响乳腺癌发病率的因素非常复杂,主要包括更年期,家族遗传,饮食,肥胖和过量摄入外源雌激素。乳腺癌是一种高度异质性疾病,细胞内抑癌基因和原癌基因的失衡是引起肿瘤的主要原因。尽管许多基因与乳腺癌的发生和发展有关,但正确的乳腺癌早期精准诊断和预后评估仍然非常困难。随着微阵列技术和生物信息学分析的迅猛发展,基因表达综合(Gene Expression Omnibus,GEO)数据库(https://www.ncbi.nlm.nih.gov/)中丰富的信息成为了发现差异表达基因(Differentially Expressed Genes,DEG)及其通路的重要途径[2]。因此,本项目从GEO数据库下载并分析数据集,鉴定乳腺癌中的DEG,构建蛋白质-蛋白质相互作用(protein-protein interaction,PPI)网络分析,从DEG中识别出TOP10枢纽基因,GO功能分析和KEGG通路分析枢纽基因的功能和通路。使用Human Protein Atlas、UALCAN以及Kaplan-Meier plotter数据库来研究筛选基因在乳腺癌预后中的作用,并实验验证筛选基因在正常乳腺细胞和乳腺癌细胞中的差异表达,确定乳腺癌的生物标志物。
1 材料与方法
1.1 数据采集通过GEO数据库(http://www.ncbi.nlm.nih.gov/geo/)获取3个乳腺癌的基因表达数据集,分别为GSE42568、GSE45827和GSE15852。GSE42568包含104例乳腺癌样本和17例正常样本。GSE45827包含了142例乳腺癌样本和13例正常样本。GSE15852包含了43例乳腺癌样本和43例正常样本。
1.2 筛选差异表达基因首先在NCBI中(https://www.ncbi.nlm.nih.gov/genome/)加载上述3个乳腺癌数据集,并通过GEO2R分析并下载上述3个乳腺癌数据集。随后对差异基因进行筛选,筛选标准为|log2FC|>1且P<0.05。
1.3 PPI网络构建及筛选枢纽基因通过将差异表达基因列表导入STRING数据库的方式,评估差异表达基因之间的相互作用关系,设定信度为0.15;随后,将数据导入Cytoscape软件,构建PPI网络,最终使用CytoHubba模块计算网络中每一个基因的最大团中心性分数,根据得分记前10的基因作为枢纽基因[8-10]。
1.4 肿瘤样本差异表达基因的功能通过DAVID进行GO和KEGG分析。将P<0.05作为筛选标准。
1.5 枢纽基因的验证通过UALCAN(http://ualcan.path.uab.edu)和HPA(https://www.proteinatlas.org)数据库分析mRNA和蛋白表达水平,Kaplan-Meier plotter数据库对其进行生存曲线分析。
1.6 细胞系RNA的提取及实时荧光定量PCR检测4种细胞系:人乳腺癌细胞MCF-7、MDA-MB-231、SKBR3和人正常乳腺上皮细胞MCF-10A(购自武汉普利莱)。RPMI-1640培养基购自美国Gibco BRL公司;胎牛血清购自美国Gibco BRL公司;双抗购自上海碧云天生物技术有限公司。培养液:血清:双抗比例为100∶10∶1,每T25培养瓶中加入5 mL培养液进行细胞培养,待细胞生长达到80%左右时,1 mL胰酶37 ℃消化3 min,加入培养液终止消化获取细胞进行总RNA的提取。TRizol试剂盒提取总RNA(Invitrogen公司),以3.5 μg RNA为模板, M-MLV Reverse Transcription Kit试剂盒(TaKaRa公司)配制反转录反应液,进行反转录。实时荧光定量PCR:50 μL体系,每份样品做3个复孔,取平均值;以β-actin为内参,内参引物序列为β-actin F:5′-TGACGTGGACATCCGCAAAG-3′,β-actin R:5′-CTGGAAGGTGGACAGCGAGG-3′;HMMR引物序列为HMMR F:5′-GGA GTC TTG CTC TGT GCT CTG TTG-3′,HMMR R:5′-GAG GCA GGA GAA TCG CTT GAA CC-3′,mRNA的表达水平以2-ΔΔCt表示,并进行乳腺癌细胞系和正常乳腺细胞系基因表达差异性分析。
1.7 统计学分析数据分析采用SPSS 18.0统计软件分析。计量资料以“均数±标准差”表示,用t检验分析,P<0.05为差异有统计学意义。
2 结果
2.1 筛选差异表达基因通过TBtools绘制火山图确定上、下调差异表达基因,设定|log2FC|>1且P<0.05为筛选条件。结果显示,GSE15852、GSE42568和GSE45827分别得到差异表达基因377个、5358个、6344个;其中GSE15852上调基因为244个,下调基因为133个;GSE42568中上调基因为805个,下调基因为2553个;GSE45827中上调基因为1677个,下调基因为4667个(见图1A~图C)。然后利用联川生物云平台(https://www.omicstudio.cn/tool?order=complex)绘制venn图,筛选得到了217个共同差异基因(见图1D)。

图1 3个乳腺癌数据集差异基因表达火山图及Venn图
2.2 PPI 网络构建及乳腺癌中枢纽基因的鉴定将差异表达基因列表上传至STRING(信度0.4)判断相互作用是否有意义的标准,构建了PPI网络(图2)。使用Cytoscape中的插件Cytohubba,通过MCC法对其差异表达基因进行评分,其中评分前10的基因为CDK1、TOP2A、AURKA、RRM2、MCM4、PRC1、HMMR、SMC4、RAD51AP1、GINS2(图3)。并对10个关键基因进行富集分析。
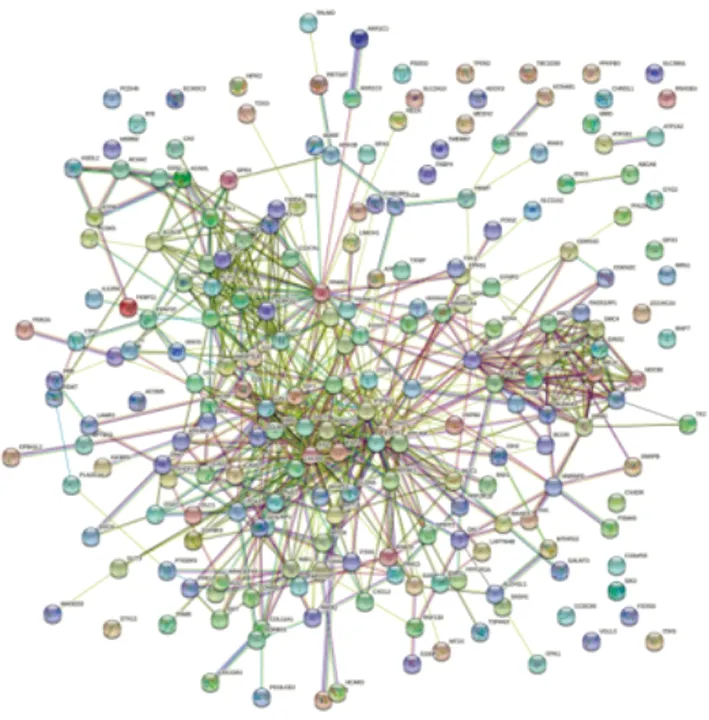
图2 差异表达基因的 PPI 网络

图3 通过MCC法得到的10个枢纽基因及其相互作用图
2.3 乳腺癌中枢纽基因的功能分析使用DAVID分析了评分TOP10的枢纽基因在乳腺癌中潜在的功能和相关机制。KEGG路径分析表明,这10个关键基因参与了p53信号通路(图4A)。10个关键基因的GO主要包括分子功能(Molecular Function,MF),细胞成分(Cellular Components,CC)和生物过程(Biological Processes,BP)。GO分析表明,TOP10的枢纽基因的MF主要为"蛋白质结合”(图4B);CC为“核原生质”“纺锤体微管”“细胞核”“细胞中间体”(图4C);主要参与的BP为“DNA复制”“有丝分裂细胞周期的G1/S转变”“有丝分裂细胞周期的G2/M转变”“DNA解旋参与DNA复制”(图D)。细胞周期内有2个重要的阶段:G1期到S期和G2期到M期,这2个过渡期正处于活跃而复杂的分子水平变化期,信号转导途径相关的调节基因可以调控周期中细胞的增殖、分裂与分化。因此,与有丝分裂细胞周期的G2/M转变显著相关CDK1,AURKA,和HMMR特别值得关注。在乳腺癌中,关于CDK1和AURKA的研究有许多,但是关于HMMR的却很少,因此我们随后专注对乳腺癌中HMMR的研究。
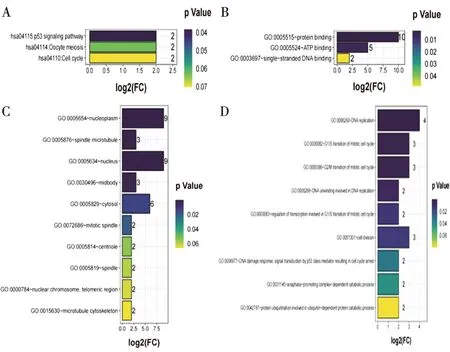
图4 TOP10枢纽基因 GO 功能分析和 KEGG 通路分析
2.4 乳腺癌中HMMR的差异性表达及生存分析为了研究HMMR在乳腺癌患者中的预后和治疗价值,通过UALCAN(http://ualcan.path.uab.edu)和HPA(https://www.proteinatlas.org)数据库分析了mRNA和蛋白表达水平,Kaplan-Meier plotter数据库对其进行生存分析。通过UALCAN检测乳腺癌中HMMR的mRNA表达水平,结果显示,与正常样品相比,乳腺癌组织中HMMR mRNA表达水平显着上调(图5A),并且HMMR的mRNA表达与患者的个体癌症分期显着相关,与第一期相比,处于癌症第二期和第三期的患者倾向于较高的HMMR mRNA表达(图5B)。进一步通过HPA数据库分析乳腺癌癌中HMMR的蛋白表达水平,结果显示,与正常样品相比,乳腺癌组织中HMMR蛋白表达水平显着上调(图5C)。Kaplan-Meier plotter数据库结果显示,HMMR的高表达与患者的总生存时间负相关(图5D)。

图5 HPA,UALCAN和Kaplan-Meier plotter数据库中对HMMR进行分析
2.5 HMMR在乳腺癌细胞系中的表达分别提取人乳腺癌细胞MCF-7、MDA-MB-231、SKBR3和人正常乳腺上皮细胞MCF-10A四种细胞系的总RNA,进行反转录,QRT-PCR检测HMMR在乳腺癌细胞中的表达水平。如图6结果所示,与正常乳腺细胞MCF-10A相比,HMMR mRNA在乳腺癌细胞MCF-7、MDA-MB-231、SKBR3的表达水平明显上调(P<0.05)。

图6 QRT-PCR检测HMMR在人乳腺癌细胞和正常乳腺细胞中表达结果图
3 讨论
在本项研究中,提取三个乳腺癌数据集数据。然后利用联川生物绘制venn图,筛选得到了217个的差异表达基因。使用STRING数据库构建PPI网络,并使用Cytoscape识别关键基因。从PPI网络的模块中筛选了前10个关键基因,分别为CDK1、TOP2A、AURKA、RRM2、MCM4、PRC1、HMMR、SMC4、RAD51AP1、GINS2。随后DAVID使用分析了评分TOP10的枢纽基因在乳腺癌中潜在的功能和相关机制。结果显示,CDK1,AURKA,和HMMR与有丝分裂细胞周期的G2/M转变显着相关。细胞周期内有2个重要的阶段:G1期到S期和G2期到M期,这2个过渡期正处于活跃而复杂的分子水平变化期,信号转导途径相关的调节基因可以调控周期中细胞的增殖、分裂与分化。因此,与有丝分裂细胞周期的G2/M转变显著相关CDK1,AURKA,和HMMR特别值得关注。在乳腺癌中,关于CDK1和AURKA的研究有许多[3-4],但是关于HMMR的却很少,因此我们随后专注对乳腺癌中HMMR的分析。
HMMR作为以透明质酸为底物,与微管蛋白互相作用的细胞外蛋白,调节细胞运动,细胞分裂,血管的生成等[5]。文献报道,HMMR的表达对肿瘤的发生、发展及预后关系并不一致,如MELE等人[6]研究发现,HMMR的过表达,促进结直肠癌细胞的生长,侵袭。而在肺腺癌细胞中的HMMR的表达水平的下调,抑制肺腺癌细胞的迁移能力[7],另外,也有一些实验研究表明,卵巢癌的进展与HMMR的过表达也存在相关性[8]。HMMR可以通过GFβ/Smad2影响胃癌患者化疗的敏感性[9]。我们通过HPA,UALCAN和Kaplan-Meier plotter数据库来研究HMMR在乳腺癌中的表达以及生存分析。结果显示,HMMR在乳腺癌中表达上调,并且与临床分期以及生存率负相关。同时,细胞实验研究结果也显示,与正常乳腺细胞MCF-10A相比,HMMR 在MCF-7、MDA-MB-231、SKBR3细胞系的mRNA表达水平明显上调。此外,我们利用蛋白质-蛋白质相互作用网络来研究与HMMR相作蛋白,通过KEGG和GO对HMMR进行通路和生物学分析,结果显示,HMMR参与微管细胞骨架构成,涉及细胞周期调控,这可能是HMMR参与调控肿瘤进展的机制。
综上所述,生物信息学和细胞实验鉴定了与细胞周期相关的HMMR在乳腺癌中过表达,HMMR的过表达对于乳腺癌患者的病程产生影响,而且伴较差的生存期。HMMR可能成为乳腺癌的预后生物标志物和潜在治疗靶点,值得进一步探讨作用的分子机制。
