KLF7对创伤性脑损伤海马神经元细胞模型凋亡和增殖的影响
2022-07-13李文媛王晓宇吕忠孝
马 多,李文媛,王晓宇,喻 静,吕忠孝,王 莹
(1.牡丹江医学院神经组织工程研究所,黑龙江 牡丹江 157011;2.厦门医学院附属第二医院,福建 厦门 361000)
创伤性脑损伤(traumatic brain injury,TBI)是目前导致死亡和致残的主要原因之一[1]。研究表明约50%以上创伤相关死亡与TBI密切相关,美国约40%TBI损伤幸存者发展为长期残疾,可导致显著的海马萎缩,并伴有语言记忆和认知功能受损。其复杂的病理生理过程涉及创伤性机械力,导致内皮或脑实质的原发性或继发性脑损伤[2]。继发性损伤发生在原发性损伤介导的神经炎症、免疫反应、氧化应激、线粒体功能障碍和细胞凋亡之后,可能持续数小时到数天。此外继发性损伤阻碍现存神经保护和修复机制,能够导致长期认知和功能性神经损伤[3]。
Krüpple样因子(Krüppel-like Factor,KLF)是一个由17个成员组成的锌指转录因子家族,调控多种重要的生理和病理过程,包括增殖、分化和细胞死亡[4]。在所有17个KLF家族成员中,KLF6和KLF7都是视神经轴突再生所必需的调控转录因子,而且,KLF7促进神经突起生长的能力是KLF6的两倍以上[5]。KLF7在成人组织中广泛表达,在脑和脊髓中占优势。在TBI模型中,一些KLF家族成员如KLF4、KLF11和KLF6在神经元损伤、缺氧/复氧损伤、轴突生长和运动神经元存活中起重要作用[6]。前期研究发现KLF7在周围神经损伤或脊髓损伤模型中可提高移植种子细胞的存活率,促进轴突再生[7]。但KLF7对TBI的机制研究尚未见报道。
本研究对HT22海马神经元细胞进行拉伸(stretch)结合氧葡萄糖剥夺(oxygen-glucose deprivation,OGD)处理,复制体外TBI细胞损伤模型[8],探讨KLF7对TBI诱导HT22细胞增殖、凋亡损伤作用及其机制,为临床TBI损伤提供新的治疗靶点。
1 材料与方法
1.1 海马神经元细胞培养及体外损伤模型的建立HT22海马神经元细胞系购自中国科学院细胞库。在10%胎牛血清(FBS)和1%青霉素/链霉素的Dulbecco培养基(DMEM),在5% CO2、37 ℃细胞培养箱中培养。每2 d更换培养基。将细胞接种在6孔板(2×107细胞/孔)上进行后续实验。
应用stretch结合OGD制备体外TBI细胞模型[9]。首先将细胞进行双轴拉伸损伤模型。HT22细胞加入涂有I型胶原6孔板上培养,然后用细胞损伤控制器(CIC)II(FlexCell International公司)处理。延迟设置为50 ms,调节器压力设置为35 psi,峰值压力设置为5.6 psi[10]。设置参数相当于中度拉伸损伤。细胞受到中等程度拉伸损伤4 h。随后细胞进行OGD处理,原培养基被缺乏D-葡萄糖的DMEM培养基替换培养,在缺氧培养箱中培养4 h,然后在进行实验前再复氧20 h。对照组HT22细胞在相同条件下处理,但不接受stretch和OGD处理。
1.2 AG490预处理及AAV-KLF7对HT22细胞的转导将HT22细胞接种于6孔板(2×107细胞/孔)上,细胞分为:control组(未处理细胞);stretch+OGD+AAV-NC(SOAN)组:由AAV-NC转染细胞24 h后,细胞拉伸后24 h进行OGD处理;stretch+OGD+AAV-KLF7(SOAK)组:AAV-KLF7转染细胞24 h后,细胞拉伸后24 h进行OGD处理。stretch+OGD+AAV-KLF7+AG490(SOAK-AG490)组:首先JAK2/STAT3抑制剂AG490预处理细胞30 min,再经AAV-KLF7转染24 h后进行细胞拉伸和OGD。sretch+OGD+AAV-NC(SOAN-AG490)组:细胞经AG490预处理30 min后,再经AAV-NC转染24 h后进行牵张和OGD。
在AAV转染、细胞拉伸和OGD之前进行AG490预处理[11]。SOAK+AG490和SOAN+AG490组中AG490(5 mM的1 μL 50%乙醇,sigma公司)。在细胞拉伸和OGD之前进行AAV转染。AAV-NC(6.5×109病毒粒/mL,Vector Biolabs公司)或AAV-KLF7(6.5×109病毒粒/mL,Vector Biolabs公司),浓度为150 μL/孔[12]。
1.3 Western blot法每组细胞通过SDS-聚丙烯酰胺凝胶电泳(SDS-PAGE)和聚偏二氟乙烯(PVDF)膜转移对蛋白质样品(20 μg)进行取样和分离。加入一抗KLF7(兔多克隆IgG;1∶500)、Bcl-2(兔多克隆IgG;1∶500)、Bax(兔多克隆IgG;1∶500)、Cleaved Caspase-3(兔多克隆IgG;1∶500)、p-JAK2(兔多克隆IgG;1∶500)、total-JAK2(兔多克隆IgG;1∶500)、p-STAT3(兔多克隆IgG;1∶500)、total-STAT3(兔重组多克隆;1∶500)、GAPDH(兔多克隆IgG;1∶500)4 ℃孵育过夜。加入辣根过氧化物酶标记二抗(1∶5000)室温下孵育1 h。所有抗体均购自Thermoscitific公司。使用增强化学发光(ECL)试剂盒(GE Healthcare公司)对标记的蛋白质进行显色,使用Image J 1.42q软件进行分析。
1.4 实时荧光定量PCR(Real-time quantitative PCR,qRT-PCR)应用Trizol试剂(美国Life Technologies公司)使HT22细胞均质化,将纯化RNA稀释500 ng/μL根据cDNA生成说明,将3 μL纯化稀释RNA与PrimeScript©RT试剂混合物孵育。各引物序列为:KLF7上游5′-TTTCCTGGCAGTCATCTGCAC-3′,下游5′-GGGTCTGTTTGTTTGTCAGTCTGTC-3′;β-actin 上游5′- CCCATCTATGAGGGTTACGC -3′,下游5′- TTTAATGTCACGCACGATTTC -3′。利用2- △△ CT方法分析。
1.5 免疫荧光染色应用一抗KLF7(兔多克隆IgG,1∶200),DAPI(1∶200)和β-Ⅲ-tubulin(兔多克隆lgG;1∶200)4 ℃孵育过夜,孵育二抗FITC结合抗兔(1∶200;山羊多克隆IgG)或Alexa555结合驴抗兔二级抗体(1∶200;驴多克隆IgG),所有抗体均购自Sigma-Aldrich公司,使用Olympus BX41荧光显微镜拍摄图像,MetaMorph软件分析荧光强度。
1.6 乳酸脱氢酶(LDH)测定LDH细胞毒性分析试剂盒(Sigma-Aldrich公司)检测LDH酶活性。与实验处理的HT22细胞在37 ℃培养30 min。应用终止液(1 M HCl,50 μL)停止反应。490 nm处检测光密度值(optical density,OD)。细胞毒性(%)=(实验组OD 值/control组OD值)×100%。
1.7 Caspase-3活性测定Caspase-3细胞活性检测试剂盒(美国Sigma-Aldrich公司)检测促凋亡蛋白Caspase-3活性。细胞裂解后将5 μL 4 mM DEVD-p-NA底物、50 μL反应缓冲液与细胞上清液(50 μL/孔)37 ℃孵育2 h。使用微孔板读取器(Bio-Rad)在405 nm测定光密度值(ODs)。Caspase-3活性(%)=(实验组OD值/control组OD值)×100%。
1.8 免疫沉淀(Chromatin Immunoprecipitation,ChIP)在OGD和细胞拉伸后,收集细胞在裂解缓冲液(2 mM EDTA、300 mM NaCl、20 mM Tris-HCl、1%nonidet P-40)提取蛋白[13]。500 μg蛋白加入一抗KLF7(2 μg,小鼠单克隆IgG,Santa Cruz Biotechnology公司)及p-STAT3(2 μg,兔多克隆IgG,ThermoScientific公司)孵育过夜,用50 μL protein A/G-加琼脂糖(Santa Cruz Biotechnology公司)孵育4 h。免疫沉淀反应后,加入SDS缓冲液。然后进行上述western blot步骤分析结合蛋白。
1.9 碘化丙啶(Propidium Iodide,PI)染色应用DAPI(5 μg/mL,Sigma-Aldrich公司)在37 ℃下加入细胞15 min标记细胞核。然后添加5 μg/mL 碘化丙啶(PI)(Sigma-Aldrich公司)10 min,应用磷酸盐缓冲盐溶液(phosphate buffered saline,PBS)洗涤。将样品在4%多聚甲醛(Paraformaldehyde solution,PFA)固定10 min,Olympus荧光显微镜拍摄图像。应用ImageJ软件定量细胞活力(%)=(PI阳性/Hoechst阳性细胞)×100%。
1.10 细胞计数试剂盒-8(Cell Counting Kit-8,CCK-8)分析应用CCK-8细胞计数试剂盒检测细胞增殖和细胞毒性。每组细胞放入96孔板在37 ℃下加入CCK-8溶液(10 μL/孔)培养2 h。用微孔板读取器(Bio-Rad公司)在450 nm处测量光密度值(ODs)。细胞活性(%)=(实验组OD 值/control组OD值)×100%。
1.11 统计学分析所得数据应用SPSS 13.0软件行统计学分析,计量数据以“均值±标准差”表示。采用单因素方差分析(one-way ANOVA)和SNK法比较,P<0.05为差异有统计学意义。
2 结果
2.1 AAV-KLF7转染HT22细胞后KLF7表达与control组比较,stretch+OGD+AAV-NC(SOAN)组和stretch+OGD+AAV-KLF7(SOAK)组KLF7蛋白和mRNA表达均显著增高,而SOAK组KLF7蛋白和mRNA表达均显著高于SOAN组(蛋白,F2,6=88.10,P<0.001;mRNA,F2,6=59.75,P<0.001)(见图1)。
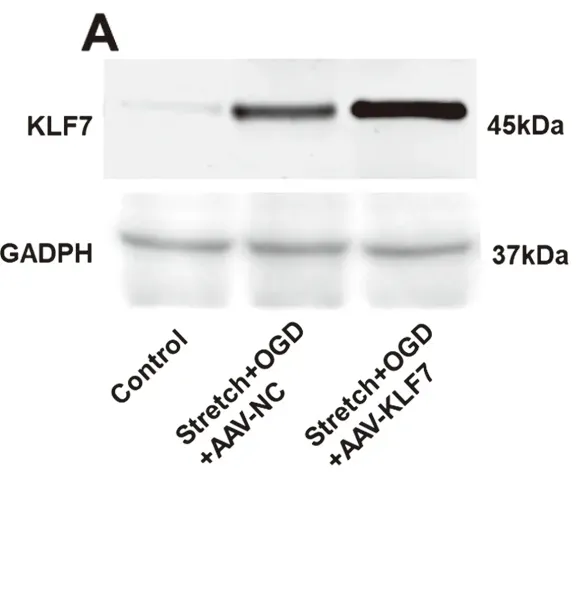
图1 AAV-KLF7转染HT22细胞后KLF7表达
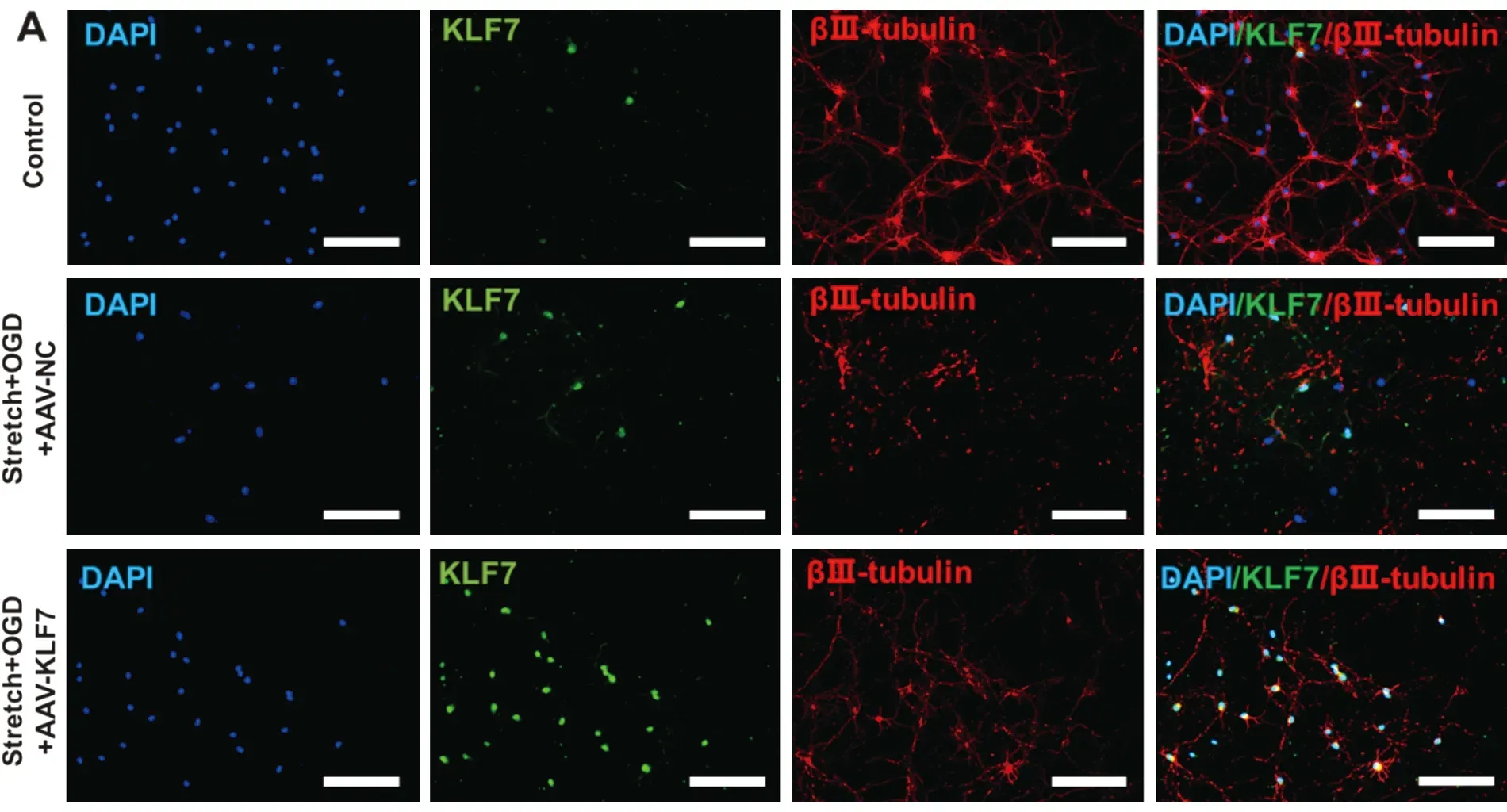
2.2 细胞免疫荧光染色检测stretch+OGD+AAV-NC(SOAN)组和stretch+OGD+AAV-KLF7(SOAK)组KLF7与βⅢ-tubulin表达均显著高于control组,而SOAK组KLF7与βⅢ-tubulin表达均显著高于SOAN组(KLF7:F2,15=264.0,P<0.001;βⅢ-tubulin:F2,15=126.3,P<0.001)(图2)。
2.3 KLF7对海马神经元凋亡的影响stretch+OGD+AAV-KLF7(SOAK)组和stretch+OGD+AAV-NC(SOAN)组PI阳性细胞比率、LDH活性及Caspase3活性显著高于control组。而与SOAN组比较,SOAK组显著降低PI阳性细胞比率、LDH活性及Caspase3活性(PI阳性细胞比率:F2,15=75.71,P<0.001;LDH:F2,15=62.45,P<0.001;Caspase3:F2,15=51.14,P<0.001)(图3A-F)。与stretch + OGD损伤组比较,control组细胞活力显著增高,而SOAK组细胞活力显著高于SOAN组(F2,15=43.06,P<0.001)(图3G)。
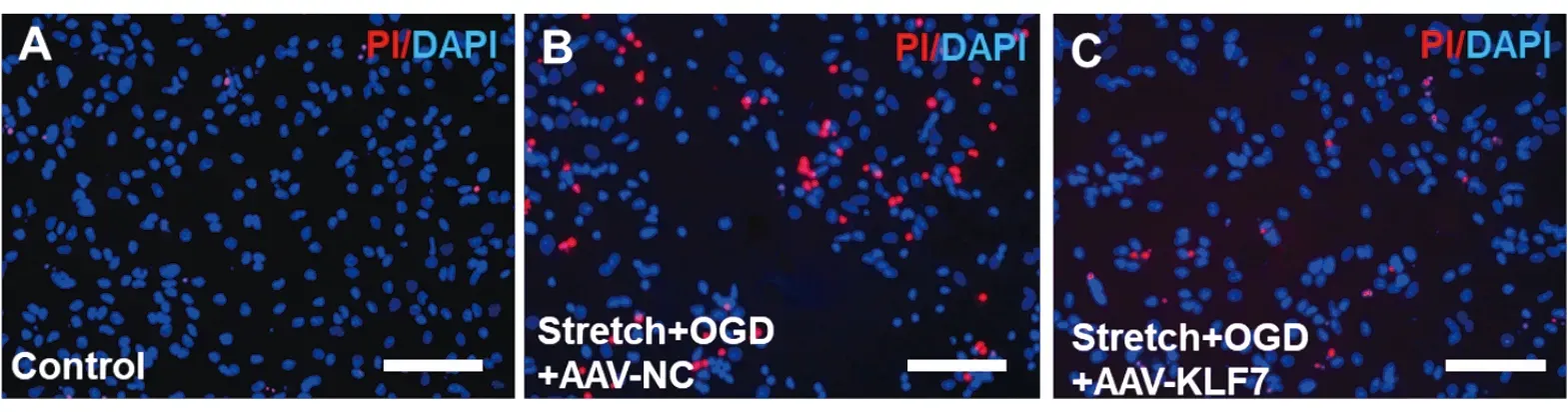
图3 KLF7对海马神经元凋亡和细胞活性的影响
2.4 KLF7调控细胞损伤的机制stretch+OGD+AAV-NC(SOAN)组和stretch+OGD+AAV-KLF7(SOAK)组p-JAK2/t-JAK2和p-STAT3/t-STAT3比值显著高于control组,而SOAK组p-JAK2/t-JAK2和p-STAT3/t-STAT3比值均显著高于SOAN组,然而与SOAN和SOAK组比较,SOAK-AG490组显著降低p-STAT3/t-STAT3和p-JAK2/t-JAK2比值(p-JAK2/t-JAK2:F3,8=139.8,P<0.001;p-STAT3/t-STAT3:F3,8=73.96,P<0.001(图4A-C)。ChIP结果表明KLF7可直接结合p-STAT3(图4D)。

图4 KLF7靶向调控JAK2/STAT3信号通路
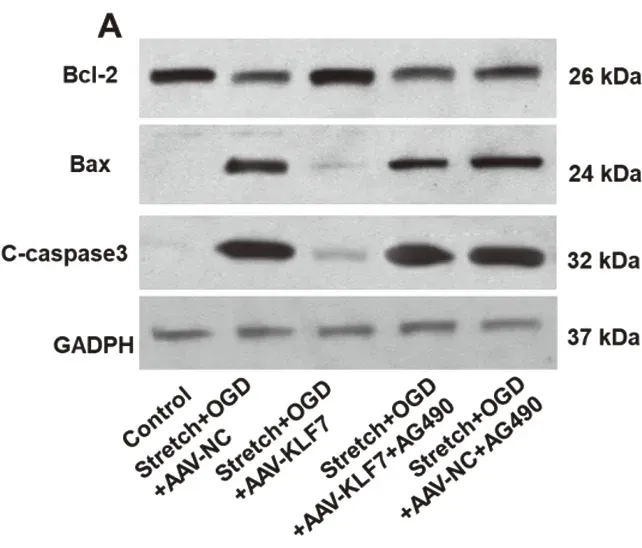
2.5 AG490对凋亡因子的影响western blot分析结果表明与control组比较,stretch+OGD+AAV-NC(SOAN)组和stretch+OGD+AAV-KLF7(SOAK)组抗凋亡因子Bcl-2蛋白表达显著降低,Bax和C-caspase3蛋白表达显著增高,然而与SOAN组比较,SOAK组Bcl-2蛋白表达显著增高,Bax和C-caspase3蛋白表达显著降低。此外,与SOAK组比较,AG490处理组显著降低Bcl-2蛋白表达,增加Bax和C-caspase3蛋白表达。其中SOAK-AG490组与SOAN-AG490组Bcl-2、Bax及C-caspase3蛋白表达无显著性差异(Bcl-2:F4,10=43.34,P<0.001;Bax:F4,10=173.6,P<0.001;C-caspase3:F4,10=93.55,P<0.001)(图5A-D)。
2.6 AG490对细胞凋亡及活性的影响与control组比较,SOAN组和SOAK组PI阳性细胞比率、LDH活性及Caspase3活性显著增高,细胞活性显著降低。然而与SOAN组比较,SOAK组显著降低PI阳性细胞比率、LDH活性及Caspase3活性,显著增高细胞活性。此外与SOAK组比较,SOAK-AG490组PI阳性细胞百分率、LDH和caspase3活性显著增加,细胞活力显著降低。其中SOAN组、SOAK-AG490组和SOAN-AG490组PI阳性细胞百分率、LDH活性、caspase3活性和细胞活性无显著性差异(PI阳性细胞:F4,15=25.07,P<0.001;LDH:F4,15=42.45,P<0.001;caspase-3:F4,15=49.83,P<0.001;细胞活力:F4,15=32.64,P<0.001(图5A~5E)。

图6 AG490对细胞凋亡及细胞活性的影响
3 讨论
创伤性脑损伤(TBI)主要由跌倒、机动车事故和娱乐活动引起,每年发病率约为190万人。目前全世界约有5300万人因一次或多次TBI相关伤害而需要医疗援助[1]。TBI发生在大脑暴露于外力时,这些外力会导致局部或弥漫性损伤,包括轴突断裂、水肿、血管损伤和神经元死亡。TBI造成的伤害从轻微到严重不等。TBI后主要损伤通常会导致继发性损伤,由炎症、氧自由基形成、钙释放和线粒体功能障碍引起的神经元死亡[2-3]。迄今为止TBI研究尚未转化为临床治疗方法。治疗TBI一种方法可能涉及转录因子的调节,对其进行适度调控可能对细胞存活产生显著影响。
前期研究表明JAK-STAT3通路介导细胞损伤调节效应[11]。JAK/STAT通路参与各种中枢神经系统疾病的进展,包括脑缺血、TBI和脊髓损伤[14]。有研究表明KLF6和STAT3共同占据促再生基因的调控DNA[15]。特别是KLF6和-KLF7通过相似的转录靶点起作用,并且共享相同的DNA结合域,这表明其具有相似基因组靶点[5]。上述结果增加了KLF7也与JAK2/STAT3信号相关的可能性,由于大量证据支持KLF7对神经系统损伤后轴突再生和神经元存活的积极影响[7]。在Kruppel样家族中,KLF7相对KLF6已被证明能促进受损皮质脊髓束轴突再生能力。本研究应用KLF7腺相关病毒转染细胞,通过western blot和qRT-PCR证实KLF7表达显著增加,证实腺相关病毒能够有效转染KLF7至HT22细胞。牵张损伤是TBI最常用的体外模型,OGD目前用于体外缺血模型。这两种方法的结合已经被证明是成功的复制TBI继发性损伤的体外细胞模型[12]。LDH活性是细胞膜完整性的指标,因此可作为细胞毒性的指标,研究表明严重TBI后,脑脊液中LDH活性和caspase-3表达显著增高,海马神经元细胞PI阳性细胞百分比显著增高。研究表明TBI损伤处理的PC12细胞显示βⅢ-tubulin降低,这与氧化损伤和炎症及神经退行性疾病进展相关[13]。在本研究中应用拉伸和OGD模拟TBI在体外造成的损伤,结果表明拉伸和OGD损伤后PI阳性细胞百分比、LDH活性和caspase-3活性显著增高,细胞活性及βⅢ-tubulin表达均显著降低,与体内TBI在原发性和继发性损伤期间发生的损伤反应相一致,再次证实拉伸和OGD联合能够成功构建TBI体外细胞模型。
本研究结果显示拉伸和OGD诱导的细胞骨架蛋白βⅢ-tubulin被KLF7的过度表达逆转。这一现象证实KLF7可以修复拉伸和OGD引起的神经突起缩短和损伤。本研究对拉伸联合OGD后凋亡相关蛋白的研究结果与前期文献一致[8]。在TBI损伤细胞模型中,Bax和C-caspase3蛋白表达均显著增高,抗凋亡因子Bcl-2表达显著降低,而KLF7高表达显著降低C-caspase3和Bax表达,显著上调Bcl-2的表达,这一结果证实KLF7能够显著抑制海马神经元TBI损伤细胞模型凋亡。
本研究在KLF7中证实了与转录因子STAT3的相互作用。KLF7相关的KLF6在启动子中被预测含有一个功能性STAT3相互作用域[11]。此外STAT3在KLF6基因启动子中富集,并且在再生相关基因路径的调控DNA中检测到共占据[15]。本研究使用JAK2/STAT3抑制剂AG490和共免疫沉淀分析,结果显示KLF7直接与STAT3相互作用,并且表明KLF7上调与JAK2/STAT3磷酸化一致。JAK2/STAT3磷酸化与抗凋亡蛋白Bcl-2及细胞凋亡相关,这些与本研究中KLF7过度表达细胞中观察到的特征相一致。此外与前期TBI研究相似,用选择性抑制剂AG490抑制JAK2/STAT3通路能够显著减弱JAK2/STAT3激活的神经保护作用。同时在拉伸和OGD的海马神经元中观察到了TBI诱导的神经元损伤,包括细胞凋亡增加和细胞活力降低,证实KLF7通过调控JAK2/STAT3通路改善TBI诱导的海马神经元损伤。但该信号通路目前仅在细胞水平得以验证,KLF7调控JAK2/STAT3保护神经元损伤机制仍有待在动物疾病模型中进一步探讨。由于TBI病理生理的复杂性,针对单一的病理生理机制干预治疗TBI临床疗效并不理想,针对TBI多靶点、全方位神经保护调控可能是TBI的未来研究方向。
综上所述,KLF7能够抑制海马神经元TBI损伤细胞模型凋亡,机制可能与调控JAK2/STAT3信号通路表达有关。
