单原子催化剂在电化学中的研究进展
2022-06-29张宏伟赵美淇韩云虎
张宏伟,陈 雯,赵美淇,马 超,韩云虎
(1.西北工业大学柔性电子研究院,西安 710129;2.清华大学化学系,北京 100084)
能源危机所导致的环境问题是目前最受社会关注的问题之一.自工业革命以来,人类生产生活的主要能源一直是煤炭、石油和天然气等不可再生的化石能源,而社会的快速发展离不开这些化石能源的迅速消耗[1].然而,急速发展的经济和不断膨胀的人口数量造成有限且不可再生的化石能源正逐渐走向枯竭[2].同时化石能源的消耗也伴随着诸多环境问题,如大量二氧化碳等温室气体的排放对全球气候产生了不可逆转的影响,化石燃料的锐减和气候变暖的加剧使得全球对可持续能源的需求不断增加.因此,风能、太阳能等可替代传统化石能源的可持续能源的发展变得格外重要,同时也刺激了相关的可持续能源储存和转换技术的广泛研究,包括燃料电池、金属-空气电池、全解水和二氧化碳还原等[3].燃料电池、金属-空气电池和全解水等能源转换和存储技术的发展可以促进能源的高效利用,新型二氧化碳还原技术的发展可以在生产能源的同时降低大气中的二氧化碳水平,协作解决相关能源与环境问题.在这些能源储存和转换技术中,高效和高选择性电催化剂的开发是实现能量快速转换的核心,其有助于降低反应势垒、促进电化学过程的动力学[4].目前,性能最为优异的电催化剂是基于Pt,Ru,Pd和Ir等贵金属的催化剂,由于稀缺性和高成本特点,限制了其大规模的实际应用[5~7].为了解决上述难题,研究人员逐步聚焦新型高效和高选择性电催化剂的开发研究,如近期备受关注的单原子催化剂[8~12].
单原子催化剂是近年来材料领域的一种新兴材料,通过减小金属纳米颗粒的尺寸,从三维到二维至一维,直至单个原子,可以显著提高催化剂的催化活性和选择性[13].单原子催化剂中高度分散的位点不仅有利于提高各种电催化反应的催化活性和选择性,而且极大地提高了金属原子的利用率,并降低大规模应用的成本[14~16].然而,广泛报道的大多数单原子催化剂仅展现单一功能的电化学活性,难以独立满足清洁和可持续能源器件的实际应用要求.因此,发展具有多功能电化学活性的单原子催化剂逐步引起众多科研人员的研究兴趣并取得了一些进展.基于此,本文首先介绍了单原子催化剂的发展历史入手,重点总结了有关双功能单原子催化剂在电化学应用中的最新研究成果,介绍了用于电催化析氧及氧还原反应(OER-ORR)、析氢和析氧反应(HER-OER)及其它双功能电化学反应的单原子催化剂的研究进展,并介绍了上述双功能单原子催化剂的应用研究.最后,总结和展望了双功能单原子催化剂所面临的挑战和发展前景.
1 单原子催化剂
在自然界中,一些生物分子中存在作为“催化剂”的单个原子,对许多生命活动起到关键作用,如叶绿素中的Mg是许多酶的活化剂,保证了植物光合作用的顺利进行;血红素中的Fe通过与血红蛋白结合,促进了细胞的新陈代谢等[17,18].在科学研究中,最早的单个金属原子作为催化活性中心可以追溯到1995 年,Thomas 等[19]报道了一种位于介孔二氧化硅上的单Ti 原子催化剂,并用于环烯烃的环氧化反应.Iwasawa等[20]在1999年合成了分散在MgO载体上的Pt单原子,其对丙烷燃烧的活性与金属Pt颗粒相当.2000年以后,有关单原子催化剂的研究逐渐增多.如,Flytzani-Stephanopoulos 研究组[21]通过共沉淀和氰化钠浸出NPs,合成了一种用于水煤气变换反应的Au/Pt单原子催化剂.Lee等[22]在2007年报道了一种位于Al2O3上的单Pd位点催化剂,并分析了孤立Pd位点的配位环境.直到2011年,张涛院士团队[23]报道了里程碑式的研究成果—一种锚定在FeOx上的Pt单原子催化剂,其CO氧化活性比Pt团簇好很多,并首次提出“单原子催化剂”的概念.其通过实验光谱和计算也阐明了催化过程中的活性位点及其结构,该研究为实现高催化性能和减少贵金属的使用指明了研究方向.在此基础上,后续的10年间单原子催化剂的发展迅速,可以发现,发展单原子催化剂最初的目的是为了高效利用每一个贵金属原子,但是随着表征分析技术的发展以及对单原子催化机理研究的深入,单原子催化剂逐渐成为材料科学和催化领域的研究前沿,并且已经扩展到非贵金属材料,应用方向包括电催化、光催化和热催化等.
单原子催化剂是一类将孤立的金属原子均匀分散在载体上的新型催化剂,在其问世之前,负载型金属纳米结构催化剂是工业过程中应用最广泛的多相催化剂.随着研究的深入,研究人员发现金属颗粒尺寸是决定催化剂性能的关键因素,从发展之初的金属块体材料到纳米颗粒和亚纳米团簇,最后尺寸减小到单个金属原子.尺寸的减小增加了金属物种的不饱和配位环境和金属组分的表面自由能,从而使金属原子易发生团聚.因此,使用与金属原子具有强相互作用的支撑材料可以防止这种聚集发生,形成稳定、均匀分散且具有高催化活性的金属单原子催化剂.发展到如今,金属粒子尺寸已可以减小至单个原子,此时金属原子的表面自由能达到最大,导致其与载体间存在复杂的相互作用,使得单原子催化剂具备独特的化学性质[图1(A)][24].此外,Xu等[25]研究了不同负载量的Au/ZrO2催化剂对1,3-丁二烯的选择性加氢反应,催化活性结果与图1(B)中的曲线相吻合,从而确定并提出了孤立的Au原子是催化剂的真正活性位点,这也是一个单原子催化剂比纳米结构催化剂具有更好催化性能的典型例子.这是由于在催化过程中,低配位的金属原子通常作为活性位点,每个金属原子的比活性通常随金属颗粒尺寸的减小而增加,而单原子催化剂在反应中所有的金属原子都暴露在反应物中,使其具有极高的金属利用率和比活性.

Fig.1 Schematic illustrate of the changes of surface free energy and specific activity per metal atom with metal particle size and the support effects on stabilizing single atoms(A),specific activity as a function of metal loadings/sizes(B)[24]
与纳米结构催化剂相比,单原子催化剂具有众多优势:首先,单个原子和载体之间的强相互作用导致了可调节的电子结构,从而提高了其催化性能;其次,单原子催化剂在电催化反应中表现出量子尺寸效应,导致离散的能级分布和独特的最高占据分子轨道-最低未占据分子轨道间隙(HOMOLUMO)[26];再次,单原子催化剂的不饱和配位环境对活性位点上反应物的吸附和活化起到重要作用,有利于降低电化学反应的能垒[27];最后,单原子催化剂的高极性可以捕获电化学过程中产生的可溶性中间体,并抑制不利的穿梭效应[28,29].此外,单原子催化剂具有明确定义和均匀分散的单位点,在揭示催化剂的高活性和选择性机理方面具有巨大的潜力.
然而,由于单个原子具有极高的表面能、制备过程易发生原子团聚的特点,单原子催化剂的发展还面临合成难与稳定性差等问题.其应用的一个关键挑战是面对高温或苛刻的反应条件,在不影响催化活性的情况下稳定载体上的孤立金属原子[30~32].为了解决该问题,通常采用低金属负载量以尽量避免合成过程中的团聚现象.但在实际的合成和反应工况条件下,理想的单原子催化剂往往要求具有高活性、高稳定性和高负载量[33].因此,制备高负载量的单原子催化剂并实现金属原子的均匀分散仍面临严峻的挑战.
本文所讨论的具有双功能活性的单原子催化剂是指,同时表现出优异的OER-ORR,OER-HER、氢氧化反应(HOR)-ORR和二氧化碳还原反应(CO2RR)-OER等性能的原子分散的单金属位点或双金属位点催化剂.这些催化剂在过电位、动力学速度及稳定性等方面具有较优异且均衡的电化学性能,使其应用的范围更广.设计合成时,通过整合双功能特性和调节协同效应,制备具有双功能活性的单原子催化剂,来提高电催化活性是一种极具前景的策略,具有双功能活性的单原子催化剂常用的设计方法有浸渍共沉淀法、空间限域法、化学刻蚀法和配位环境调控策略.如,Zhao等[34]采用点击限制策略,预先构建一个稳定的配位环境,之后根据点击化学的原理,通过共价键将得到的分子进一步锚定到基底上合成了M-N-C SAC,显著提高了双功能氧电催化剂性能.一方面,共价键的固有方向性和饱和性确保了接枝的过渡金属分子的单分散性;另一方面,与范德华力相比,共价键的相互作用相对较强,阻止了热解过程中过渡金属原子的聚集趋势.Chen 等[35]采用空间限制策略,通过调节LDH 的层间阴离子(如等)和M2+/M3+阳离子的比例,合成了一系列数量可调的具有独特交互协同作用的双功能单原子催化剂.其中,层间Pt单原子极大地提高了Ni3FeLDH载体的电子转移能力,增加了Ni的价态并缩短了Ni/Fe—O键键长,使催化剂获得了优异的HER及OER活性.
2 OER-ORR双功能单原子催化剂
近年来,金属-空气电池作为正在迅速发展的一类能源存储装置,OER 和ORR 是其不可或缺的核心反应.其通常由锌阳极、膜分离器、集流体和含有双功能氧电催化剂的空气阴极所组成,其电解质大多数为碱性或中性.
放电过程具体的电化学反应如下:
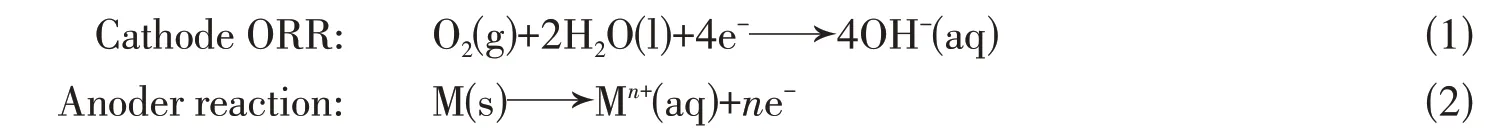
充电过程具体的电化学反应如下:
OER和ORR过程都是相当缓慢的动力学过程,涉及一系列复杂的中间反应和多步电子转移路径,使得反应过程中需要采用高效的电催化剂.然而,现有的大多数氧电催化剂仍存在一些亟需解决的问题,如,ORR 起始电位和半波电位较低、OER 的过电位较高,且难以在同一催化剂中同时实现优异OER 和ORR 双功能活性的平衡[36,37].目前,许多研究人员已经投入了大量精力研究高效的氧电催化剂,以促进金属-空气电池的发展和应用.鉴于单原子催化剂的快速发展,研究人员发现可通过合理设计,制备出具有优异的OER和ORR活性的双功能单原子催化剂,以此作为解决制约金属-空气电池大规模应用问题的关键途径.基于此,目前许多具有高活性的OER 和ORR 双功能单原子催化剂已经被开发出来,下面将对这些单原子催化剂进行详细介绍.
在电催化OER 过程中,Co 基催化剂具有较大的优势和良好的发展前景,因此,在报道的OER 和ORR双功能单原子催化剂中,Co基单原子催化剂占比较大[38].单原子M-N-C催化剂(M是过渡金属)由于超高的本征反应活性、最大化的原子效率、碳骨架所赋予的良好的电子传导和离子传输速率等优点,使其具有优异的电化学性能[39~41].Li 等[42]设计了一种与石墨烯组合的钴配位卟啉框架杂化物(Co-G@POF)作为前驱体,热解制备了Co-POC单原子电催化剂.选择钴配位卟啉框架(Co-POF),是由于其具有类似Co-Nx-C 的分子结构和易于合成的优点[43].通过X 射线吸收近边结构光谱(XANES)发现,Co-POC 催化剂在7711 eV 处存在一个边缘峰,表明其具有Co-N4结构[图2(A)][42,44].如图2(B)所示,Co-POC催化剂的傅里叶变换扩展X射线吸收精细结构谱(EXAFS)只显示位于1.32 Å(1 Å=0.1 nm)的一个主峰,与对照的CoTPP样品在1.38 Å处的Co-N峰一致,因此将该峰确定为Co-N/C配位结构[45].此外,在0.1 mol/L KOH 溶液中评估了Co-POC 催化剂的氧电催化性能,研究发现Co-POC 催化剂与贵金属基电催化剂相比,具有相当的ORR 半波电位和OER 过电位,分别为0.83 V 和470 mV,塔菲尔斜率也显示出Co-POC催化剂具有最低的斜率,表明其具有优异的ORR-OER双功能电催化活性和快速的反应动力学[图2(C)~(F)].
单原子催化剂的合成需要理性的设计,Han等[46]通过调整双金属Zn/Co沸石咪唑框架(ZnCo-ZIFs)前驱体中锌和钴的含量,分别合成了负载在N掺杂多孔碳上的Co纳米颗粒、Co原子团簇和Co单原子等催化剂,该研究在成功制备单原子Co催化剂的同时,探讨了尺寸对于催化剂电化学活性的影响.具体而言,通过精确调节ZIF结构中的Zn/Co摩尔比,使Co金属形成不同程度的分散,研究发现Zn/Co摩尔比为0∶1时,Co原子基本没有分散,热解之后经酸洗形成了Co纳米颗粒催化剂(Co-NPs@NC),引入的锌原子可以使钴原子在热解时分离;当Zn/Co摩尔比为2∶1时,热解后形成ZnCo纳米颗粒,并在最终的酸洗后形成原子水平的Co 团簇(CoACs@NC);当Zn/Co 摩尔比为8∶1,最终得到Co 单原子催化剂(Co-SAs@NC)(图3).在这些材料中,Co-SAs@NC 催化剂的ORR 活性最好,具有0.96 V 的起始电位、0.82 V的半波电位和15.2 mA/cm2的最大动力学电流密度,接近商业贵金属Pt/C催化剂的活性(性能参数分别为0.98 V,0.82 V 和15.2 mA/cm2).此外,Co-SAs@NC 催化剂也表现出相当大的OER 活性.Yang等[47]基于之前对单原子Co-N-C催化剂双功能性质的理论预测,提出了一种表面辅助方法来合成具有原子分散Co-N4活性位点的单原子Co催化剂(CoN4/NG).如图4(A)所示,CoN4/NG催化剂的原料为g-C3N4、Co盐和F127复合前驱体,通过超声使吸附Co的F127进入g-C3N4夹层,其中F127是聚氧乙烯-聚氧丙烯-聚氧乙烯(PEO-PPO-PEO)的水溶性表面活性剂.然后,在氮气条件下对前驱体进行热解,所得碳化产物在室温下蚀刻24 h,最终得到CoN4/NG 催化剂.所制备的单原子CoN4/NG 催化剂具有优异的ORR活性,半波电位为0.87 V,比商业20%Pt/C高20 mV,此外,CoN4/NG催化剂在10 mA/cm2下对OER也表现出380 mV的低过电位[图4(B)和(C)].研究证实,CoN4/NG催化剂具有出色的ORR和OER双功能性能归因于原子分散的Co-N4活性物种.

Fig.3 Preparation of the Co‐NPs@NC,Co‐ACs@NC and Co‐SAs@NCcatalysts[46]
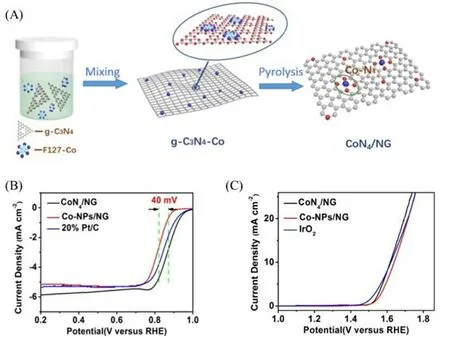
Fig.4 Schematic illustration of the synthesis of single‐atom CoN4/NG catalyst(A),RRDE ORR polarization curves of CoN4/NG,Co‐NPs/NG and 20%Pt/C in O2‐saturated 0.1 mol/L KOH solution at electrode‐rotation speed of 1600 r/min(B),OER LSV plots of CoN4/NG,Co‐NPs/NG and IrO2 in 0.1 mol/L KOH(C)[47]
Mn-Nx-C材料中Mn金属中心的多重化学态(如+2,+3和+4价态)和可控的配位结构促进了其作为电催化氧反应的应用潜力,且金属Mn具有储量丰富、价格低和环保等优点[48,49].基于此,Shang等[50]通过原子界面工程制备了一种单原子Mn 催化剂(MnSAC),催化剂中含有的孤立的Mn 位点是其优异的OER和ORR双功能催化活性的主要来源.他们以SiO2为牺牲模板、醋酸锰和壳聚糖作为原料,然后在80 ℃下搅拌直至溶液干燥,将收集的产物经研磨后放入管式炉中,在氩气条件下煅烧3 h.随后将煅烧后的产物在室温下用HF 溶液刻蚀,洗涤干燥后得到最终的MnSAC[图5(A)].由于Mn 原子与碳基底的强相互作用,使得MnSAC 不仅展现优异的ORR 和OER 活性,在碱性条件下ORR 半波电位达到0.915 V,在10 mA/cm2下的OER过电位仅为350 mV,且具有优异的稳定性.此外,同步辐射技术表征揭示了催化剂的活性中心原子构型为Mn-N2C2结构,且通过实验和理论模拟计算揭示了MnSAC的催化氧反应的机理.如图5(B)所示,Mn-N2C2结构在整个ORR过程都是放热的,表明Mn-N2C2结构具有最优的ORR 活性.理论计算得到Mn-N4,Mn-N3C1,Mn-N2C2和Mn-N1C3催化OER 的过电位分别为0.981,1.011,0.871和2.061 V,Mn-N2C2最小的过电位表明其具有最优异的OER活性.该研究表明,通过金属活性中心配位环境调控可以极大促进材料电催化氧反应的优化,助力可持续能源应用的研究.
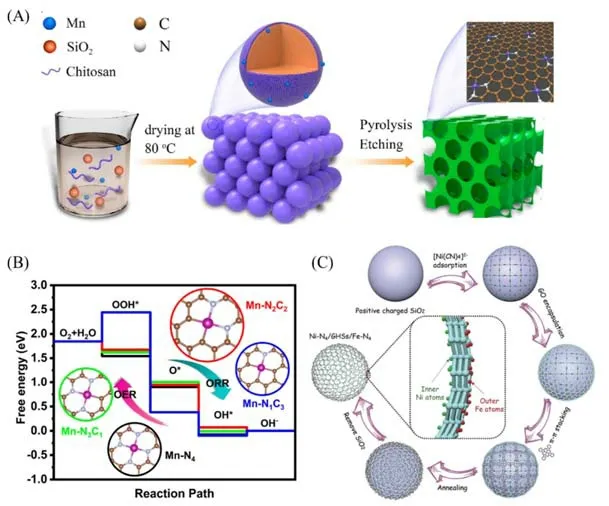
Fig.5 Illustration of the formation of MnSAC(A),free energy diagram for ORR and OER on Mn‐N2C2,Mn‐N4,Mn‐N3C1 and Mn‐N1C3(B)[50],synthetic procedure of the Ni‐N4/GHSs/Fe‐N4 catalyst(C)[51]
除了研究以单种金属为活性位点同时用作OER和ORR的活性位点外,在同一催化剂中,同时引入利于OER的金属位点和利于ORR的金属位点,构筑双金属双功能单原子催化剂也是切实可行的策略。如图5(C)所示,Chen等[51]提出了一种逐级自组装策略,开发了Ni-N4/GHSs/Fe-N4Janus材料,单Ni原子和单Fe原子分别锚定在石墨烯空心纳米球的内外壁上.经表征证实Ni和Fe单原子与4个N原子配位形成了Ni-N4或Fe-N4平面构型.研究发现,所开发的Ni-N4/GHSs/Fe-N4Janus催化剂具有优异的双功能电化学性能,位于外部的Fe-N4位点对ORR具有很高的活性,而处于内部的Ni-N4位点主要对OER起催化作用.本课题组[52]也利用发展的重力导向化学气相沉积策略合成了一种含Fe,Co单原子和FeCo纳米颗粒的双功能电催化剂.将三聚氰胺通过重力导向的化学气相沉积策略在倒置的石英片上经过Fe和Co催化生长,获得了具有碳纳米管结构的FeCo/Se-CNT催化剂,其中的Fe,Co单原子在OER和ORR催化过程中扮演了重要角色[图6(A)~(C)].FeCo/Se-CNT催化剂的ORR半波电位达到0.90 V,OER过电位仅340 mV,为其在锌-空气电池中的应用奠定了基础[图6(D)和(E)].
Fig.6 Schematic illustration of the synthesis of the FeCo/Se‐CNT catalyst(A),fourier transform EXAFS spectra of the FeCo/Se‐CNT catalyst and Fe foil(B),fourier transform EXAFS spectra of the FeCo/Se‐CNT catalyst and Co foil(C),LSV polarization curves with iR corrected for ORR in 0.1 mol/L KOH media(D),LSV polarization curves with iR corrected for OER in 1 mol/L KOH media(E)[52]
3 HER-OER双功能单原子催化剂
氢能是一种极具应用潜力的绿色能源,具有能量密度高、污染物零排放等优点,被视为碳基燃料的优良替代品[53,54].全解水制氢是一种工艺简单、能量转换效率高且环境友好的生产氢气方法[55].完整的全解水过程包含HER和OER,在不同的电解液中其两个半反应的路径有所差异:
在碱性电解液中:
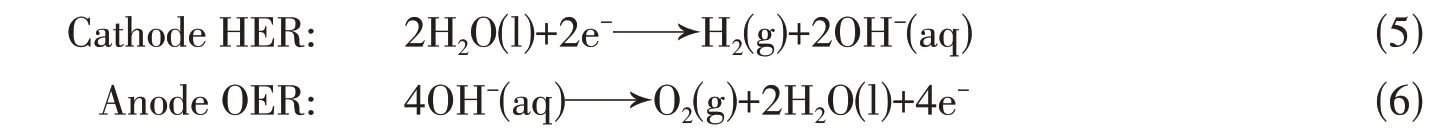
在酸性电解液中:

目前为止,以氧化物、石墨碳、硫化物及硼化物等为基底负载单分散的金属活性原子的单原子催化剂被广泛研究.Li 等[59]通过水热法结合高温煅烧制备了一种将Co 单原子掺入RuO2球体(Co-SAC/RuO2)的HER和OER双功能单原子催化剂[图7(A)].XANES谱中,Co-SAC/RuO2的CoK边吸收曲线位于CoO和Co3O4之间,接近CoO,表明Co的价态接近于+2价.傅里叶变换EXAFS谱仅显示在1.47 Å附近一个强峰,可归属于Co 原子与RuO2晶格中氧的Co-O 配位,未见类似的Co-Co 配位峰(2.17 Å)证明Co以单原子形态存在于RuO2球体中[图7(D)和(E)].电化学测试表明,该催化剂具有优异的双功能活性:在1 mol/L KOH 中OER 的过电位仅为200 mV@10 mA/cm2[图7(B)和(C)];在0.5 mol/L H2SO4中HER过电位仅为45 mV@10 mA/cm2[图7(F)和(G)].

Fig.7 Schematic illustration of the synthesis of the Co single atom incorporated RuO2 sphere(A),LSV curves(B)and overpotentials at 10 mA/cm2(C)of Co‐SAC/RuO2,RuO2 sphere,commercial RuO2 and Pt/C of HER in 0.5 mol/L H2SO4,XANES spectra(D) and FT‐EXAFS spectra(E) of Co‐SAC/RuO2,CoO,Co3O4 and Co foil,LSV curves(F)and overpotentials at 10 mA/cm2(G)of Co‐SAC/RuO2,RuO2 sphere,commercial RuO2 and Pt/C of OER in 1 mol/L KOH[59]
金属有机框架(MOFs)是由金属位点和有机配体构成的具有有序结构的金属有机化合物,因其微孔和可设计性被认为是理想的单原子催化剂的前驱材料.Dou等[60]利用沸石咪唑骨架67(ZIF-67)为前驱体合成了一系列单原子催化剂(M1@Co/NC,M1=Ir1,Pt1,Pd1,Ru1,Fe1,Ni1).ZIF-67结构中咪唑五边形的独特p电子可与金属离子键合形成强的键合,从而锚定单个金属原子,抑制聚集.此外,ZIF-67可以转化为Co纳米颗粒分散于氮掺杂多孔碳的异质载体(Co/NC),Co和NC均可以作为单原子基底,形成M1@CoO和M1@NC两种活性位点[图8(A)~(E)].电化学测试表明,Ir1@Co/NC表现出良好的HER和OER 双功能活性[图8(F)和(G)],DFT 计算表明,催化剂中Ir-NC3结构是高效HER 的活性位点,而Ir@CoO(Ir)为OER活性位点[图8(H)和(I)].此外,MXenes材料因具有活性位点丰富、高导电性和良好的结构稳定性等优势,也被作为单原子催化剂基底[61,62].Kim等[63]使用第一性原理计算及基于机器学习(ML)的描述符发现,单个Ni 或Co 原子嵌入MXenes 中时为最佳的吸附质析出机制(AEM)和提高O2p带到激活的晶格氧化机制(LOM)提供了合适的电子数量.研究证实了MXenes组合(Ni掺杂Sc3N2O2和Ni掺杂Nb3C2O2)具有优异的HER和OER双功能催化活性与稳定性.
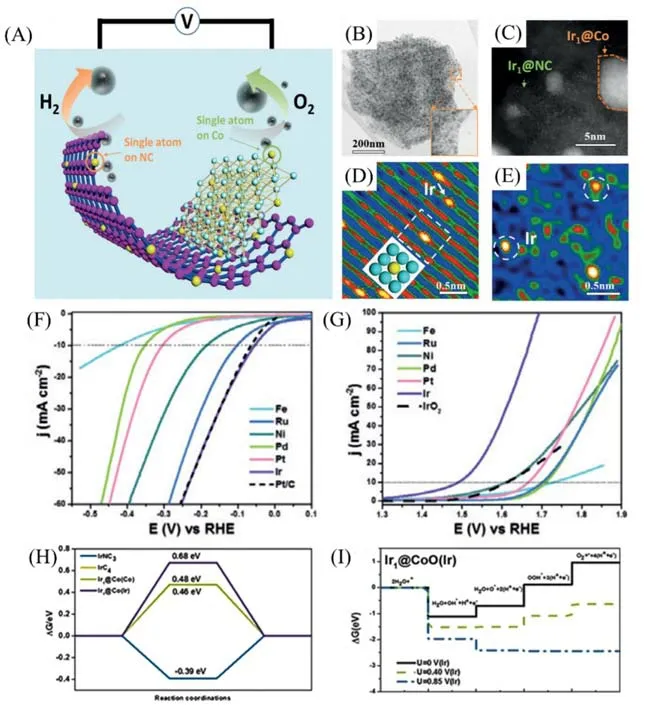
Fig.8 Illustration of the working mechanism of the prepared electrodes(A),Annular bright‐field‐STEM images of Ir1@Co/NC at low magnification(B),HAADF‐STEM image of Ir1@Co/NC at high magnifi‐cation(C),HAADF‐STEM image of Ir1@Co region(D),FFTI‐HAADF image of Ir1@NC area(E),po‐larization curves of the HER and the OER of Ir1@Co/NC,Pt1@Co/NC,Pd1@Co/NC,Ru1@Co/NC,Ni1@Co/NC,Fe1@Co/NC,commercial Pt/C and IrO2 electrodes(F,G),the free‐energy diagrams for the HER at pH=14 on IrNC3,IrC4,Ir1@Co(Co) and Ir1@Co(Ir)(H),the reaction free energies of the intermediates on Ir@CoO(Ir)(I)[60]1
资源丰沛且成本低廉的二维二硫化钼(MoS2)材料在催化领域备受关注,计算和实验研究都证明其边缘位点具有高的HER催化活性[64,65].Lee等[66]开发了一种垂直生长在三维TiN纳米棒阵列(NRs)上的Co单原子分散的MoS2纳米片(CoSAs-MoS2/TiN NRs),可以作为一种在全pH条件下进行全解水的高活性双功能电催化剂[图9(A)].Co单原子和层状MoS2之间的强相互作用调节了Co和MoS2的电子密度,为反应物分子提供了更多活性位点,并优化了全解水过程中间物种的吸附/解吸能.CoSAs-MoS2/TiN NR在酸性、碱性和中性条件下的HER过电位分别为187.5,131.9和203.4 mV@10 mA/cm2;OER过电位分别为454.9,340.6和508.0 mV@10 mA/cm2[图9(B)~(G)].其双功能活性优于此前报道的非贵金属HER和OER双功能电催化剂,并且在全pH条件下均具有良好的稳定性.Cheng等[67]使用密度泛函理论(DFT)计算,筛选了负载在MoS2边缘的28种过渡金属(TM)单原子催化剂作为全解水的双功能电催化剂的潜力.从化学环境和活性中心的局部结构导出的简单方程用作结构描述符来预测基于MoS2SAC 的OER 活性.最终发现由Pt 单原子修饰的T1-空位显示HER 和OER 反应的最低理论过电势分别仅为−0.10和0.46 V,与贵金属基全解水催化剂活性相当.此研究通过吸附-活性-结构关系证实了结构描述符在不同S配位环境下的适用性,并解释了Pt/T1-空位高催化活性的原因.

Fig.9 Schematic illustration of the fabrication of the CoSAs‐MoS2/TiN hybrid(A),iR‐corrected polariza‐tion curves of HER activity of the as‐prepared samples in 0.5 mol/L H2SO4(B),1.0 mol/L KOH(C)and 1 mol/L PBS(D),iR‐corrected polarization curves of OER activity of the as‐prepared samples in 0.5 mol/L H2SO4(E),1.0 mol/L KOH(F),and 1 mol/L PBS(G)[66]
单层硼(Borophene Monolayers,BMs)是一种质量轻、机械强度高、导电性良好的二维金属材料,其晶胞由缺电子六角孔和富电子三角孔组成,这些特殊的六角孔结构有利于吸附外部金属原子,是一种理想的单原子锚定基底材料[68,69].Wang 等[70]第一次提出计算机辅助设计的单层硼负载的双功能单原子催化剂,得到的β12单层硼负载的孤立镍原子(Ni1/β12-BM)催化剂表现出良好的HER和OER双功能活性.此外,通过动力学模拟得到β12-BM上Ni原子的扩散势垒达到1.68 eV,可以有效防止Ni原子在制备和催化过程中的聚集[图10(A)和(B)].掺杂型硼化物也可以作为单原子催化剂的基底材料,二维六方氮化硼(BN)纳米片结构类似于石墨烯,具有高热稳定性和耐腐蚀性的优点[71,72].Xu等[73]通过密度泛函理论(DFT)系统研究了以氮掺杂的缺陷氮化硼纳米带(BNNR)为基底负载TM-N4结构位点催化剂(TMN4-BNNR,TM=Cr,Mn,Fe,Co,Mo,Ru,Rh)的双功能催化活性.结果表明,所有过渡金属原子均可与N掺杂缺陷BNNR上未配位的4个N原子形成强键合,其中FeN4-BNNR和RhN4-BNNR可作为HER和OER双功能催化剂,过电位分别为0.07/0.52 V和0.18/0.27 V[图10(C)].
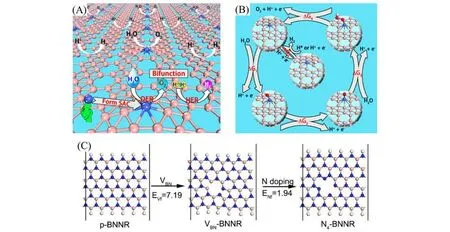
Fig.10 Schematic of the single‐atom,bifunctional catalyst for overall water splitting(A),elementary reactions of OER and HER and the structures of the adsorbed states for each species,including *H,*OH,*O,and *OOH(B)[70],the schematic illustration of proposed synthetic route of TMN4‐BNNR(C)[73]
4 其它双功能单原子催化剂
双功能单原子催化剂除了同时催化ORR 和OER,HER 和OER 性能以外,一些单原子催化剂在联氨氧化(HzOR)、二氧化碳还原(CO2RR)、乙烯析出(EER)、芳香族氯化(ACR)和氢氧化反应(HOR)等方面优良的催化性能,也被研究人员用于替代电解水制氢体系中受动力学缓慢限制的阳极OER过程,实现以更低的过电位电解制氢.因此,研究人员正在探索使用更具热力学优势的小分子氧化反应取代电解水阳极的氧析出反应.Li等[74]采用硫化和简易恒流沉积策略制备了固定在二硫化钨硫空位上的单原子Ru催化剂(WS2/RuSAs),如图11(A)所示,其可催化联氨氧化(HzOR)并用作电解水制氢的阳极和阴极催化剂,HzOR 和HER 作为阴极、阳极发生的反应实现电解水制氢,在10 mA/cm2时,展示出−74和−32.1 mV的超低电位.理论计算结果表明,RuSAs在WS2中具有关键作用,Ru位点既促进HER过程中水的解离,同时在HzOR过程中促进肼逐步脱氢.
在现有的衬底中,石墨烯或类石墨烯衬底以大比表面积、优异的电导性、高电化学稳定性和轻质等优势,使其负载的单原子催化剂显示出巨大的应用前景.通常,石墨烯或类石墨烯负载的单原子催化剂的内在活性在很大程度上取决于金属中心与反应底物之间的相互作用,包括O2、CO2及一些中间产物.为了研究3d,4d和5d轨道的不同过渡金属中心对单原子催化剂固有活性的影响,Deng 等[75]采用密度泛函理论计算,系统地探讨了O 端V2C MXene(V2CO2)负载TM 单原子催化剂作为ORR 和HOR反应催化剂的潜力(在碱性溶液中,V2CO2被O封端,具有优异的导电性和高热稳定性,电极电位高于−0.25 V(vs.SHE)[76].理论计算结果表明,TM单原子和V2CO2的结合改变了V2CO2的电子结构,丰富了活性位点,从而调节了ORR和HOR的中间体的吸附能和催化活性.如图11(B)所示,ORR物质在TM上的吸附具有相似的变化趋势,吸附强度主要取决于TM 的轨道电子数,可以推测Sc-V2CO2,Mn-V2CO2,Cu-V2CO2,Rh-V2CO2和Au-V2CO2具有中等的ΔEOH*[接近Pt(111)的0.95 eV],具有很大的ORR 催化潜力。如图11(C)所示,随着TM附近的O位被H原子覆盖,大部分ΔGH*在TM-V2CO2上趋于0,其 中Sc-V2CO2,Ti-V2CO2和V-V2CO2的ΔGH*小 于Pt-V2CO2,Cr-V2CO2,Mn-V2CO2,Fe-V2CO2和Co-V2CO2的ΔGH*与Pt较为接近.因此,Mn-V2CO2和Sc-V2CO2有望成为燃料电池双功能催化剂的最佳候选者.
精神科疾病的临床特点主要表现为:患者整个心理过程发生紊乱,绝大多数患者疾病周期长、病程迁延,较为严重者甚至出现思维紊乱、行为怪异,其工作、学习和生活自理能力均受到严重损害[7-8],护理风险管理在精神科的临床应用显得尤为重要,如何提高精神科护理质量,减少护理安全不良事件的发生应成为精神科护理的核心工作任务。
Fig.11 Schematic illustration of the proposed alkaline HER and HzOR mechanism on Sv‐WS2,Sv‐WS2@Ru NPs,and Sv‐WS2/Ru SAs(A)[74],O,OH and OOH adsorption energy(ΔEads) on TM‐V2CO2(B),HOR free energy diagrams on TM‐V2CO2 at θH of 9/16(C)[75]
MOF 衍生催化剂的结构设计和金属位点集成规律,与MOF 衍生催化剂中主导电催化性能的金属位点相关的关键催化过程息息相关.基于此,Zhao 等[77]通过X 射线光谱、电子显微技术和电化学技术,阐明了催化剂中单原子活性位点的可视识别.如图12(A)~(D)所示,通过S辅助位点纯化策略或酸洗策略完全去除金属Co颗粒后(Co-SAC,未去除金属Co颗粒的样品记为Py-ZIF),留下的单原子Co位点对ORR和HER没有显示出明显的活性下降,ORR选择性反而显著升高,这揭示单原子Co在催化过程中承担主要的活性和选择性的作用.Li等[78]提出一种光化学固相还原方法,制备了负载在氮掺杂多孔碳上的单原子Pt催化剂(Pt1/NPC),可用作ORR 和HER双功能催化剂.此外,利用这种光化学固相还原方法合成的Pt 原子可以很好地分散在碳氮载体上而没有发生团聚.Pt1/NPC 在电流密度为10 mA/cm2时,展现超高的HER活性,过电位仅为25 mV,其对ORR也展现优异的活性.
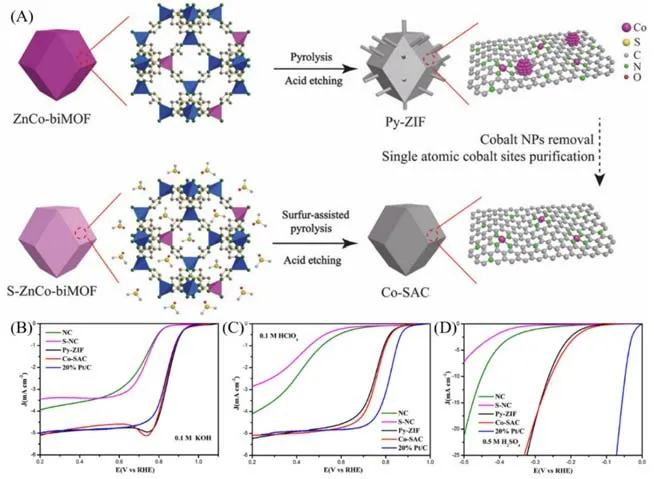
Fig.12 Illustration of the coupled evolution of single‐atom cobalt sites and metallic cobalt sites and their de‐coupling process(A),the LSV curves of catalysts NC,S‐NC,Py‐ZIF,Co‐SAC and 20%Pt/C in oxygen‐saturated 0.1 mol/L KOH(B),0.1 mol/L HClO4(C)and nitrogen‐saturated 0.5 mol/L H2SO4(D)[77]
过渡金属单原子催化剂已经被证明具有优异的CO2RR能力,生成的产物可用于生产燃料或高附加值的化学品.双金属单原子催化剂在继承单原子催化剂优点的同时,可在两种相邻的不同金属原子之间进行功能互补和协同作用,实现CO2更高效的转化.Zeng等[79]将氨基酸、醋酸铁、醋酸镍和三聚氰胺球磨混合后煅烧,然后经稀盐酸洗涤去除金属颗粒后获得NiFe 双金属单原子催化剂,该催化剂对CO2RR 和OER 均展示出优异的催化活性和稳定性.在10 mA/cm2的电流密度下,仅需电位1.54 V(vs.RHE)实现反应顺利的进行,并且在50次循环伏安测试后基本保持初始的OER活性.在饱和CO2的0.5 mol/L碳酸氢钾溶液中,NiFe双金属单原子催化剂相比于单独的Fe-SAC和Ni-SAC,前者在相同的电位范围内实现了>80%的CO法拉第效率,且在−0.8 V(vs.RHE)时达到最大(>94.5%).理论计算和实验均表明,Fe原子与相邻的Ni原子之间的轨道耦合对提高CO2RR和OER性能有极大的促进.如图13(A)所示,Hu等[80]利用周期性3×2矩形超级单体片(24个C原子)构建SACs,建立了一个含有4个吡啶N原子的空位来锚定过渡金属原子.通过密度泛函理论计算,研究了由单层Fe-N4(铁和氮共摻杂石墨烯)和M-N4(Fe/M,M 代表过渡金属原子)组成的双层单原子催化剂催化HER,ORR 和OER 的性能.从自由能和动力学角度研究发现,在一系列双层单原子催化剂中,Fe/Hf 和Ir/Fe 可分别作为催化ORR-OER 与HER 和OER 的双功能催化剂.如图13(B)和(C)所示,Fe/Hf 的ORR 的决速步骤为OH*到H2O的形成,过电位(ηORR)为0.38 V,且与IrO2催化剂(ηOER=0.60 V)相比,Fe/Hf表现出相对较低的ηOER(0.27 V).

Fig.13 Top and side views of B‐SACs(A),calculated theoretical ORR overpotential on highly active hetero‐structure systems for the ORR under U=0 V(B),calculated theoretical OER overpotential on highly active heterostructure systems for the OER under U=0 V(C)[80]
5 双功能单原子催化剂的应用
5.1 双功能OER-ORR催化剂用于金属-空气电池
金属-空气电池是一种通过金属氧化和氧气还原来产生电能的电化学电池,被认为是在新一代电子产品、交通和电能储存中应用前景极为广阔的能源设备.金属-空气电池最突出的优点是,其可将高能量密度的金属负极与具备开放结构的活性空气正极材料相结合,实现高效能源储存和转化.与商用的铅酸电池、镍-金属氢化物电池和锂离子电池相比,金属-空气电池具有更高的能量密度[81].通常研究的金属-空气电池的金属电极材料包括锂、锌、铝和镁等,其中锌电极适用于水系电解质,具有在碱性溶液中耐腐蚀、成本低和储量高等优点,因此锌-空气电池在金属-空气电池的研究中备受关注[82,83].目前,研究较多的可充电锌-空气电池可分为液态锌-空气电池和柔性固态锌-空气电池,柔性固态锌-空气电池是近年提出的一种新兴技术,是指具有特殊机械性能的可滚动、弯曲、扭曲或折叠的新型二次电池,可以满足先进电子行业与柔性电子产品对于兼容的密集电源的需要,液态与柔性固态锌-空气电池的主要区别是电解质的状态,液态锌-空气电池常用碱性电解质溶液,而柔性固态锌-空气电池则常采用碱性凝胶电解质.在锌-空气电池的充放电过程中,其核心是OER和ORR的发生.ORR和OER两种反应都是四电子过程,机制复杂且动力学反应速率缓慢,急需开发高活性氧电催化剂来促进金属-空气电池的应用发展.下面主要介绍OER和ORR双功能单原子催化剂在锌-空气电池中的应用研究.
Zhang 等[84]将FeCl3包封卟啉前驱体进行热解得到N,S 共掺杂的Fe-NSDC 催化剂,其含有孤立的Fe-Nx-C结构.受益于大量原子分散的活性位点产生高ORR和OER活性,3D导电片状结构促进离子/电子转移,大比表面积暴露了更多的活性位点,最终催化剂显示出十分优异的双功能电催化活性和稳定性.考虑到Fe-NSDC优异的双功能活性,利用其构建了液态锌-空气电池,并证明了其在实际能源器件中的可行性.研究发现,Fe-NSDC催化剂集成的可充电锌-空气电池与其它电池相比,具有1.53 V的超高开路电位、225.1 mW/cm2的峰值功率密度、在4 mA/cm2电流密度下400 次循环的稳定性、以及在100 mA/cm2下1.00 V充放电电压间隙[图14(A)~(C)].对照的Pt/C+RuO2基可充电液态锌-空气电池的失活行为,可归因于在运行过程中贵金属纳米颗粒的团聚并从碳载体中分离[85,86].Fe-NSDC独特的介孔结构不仅可以阻止催化剂活性中心的聚集,且还可以保持连续的质子和电子运动通道,从而大大提高能量效率.此外,以涂覆Fe-NSDC催化剂的泡沫镍为空气阴极、锌箔为阳极、碱性聚乙烯醇为固态电解质组装了固态锌-空气电池,卷曲状的电池显示出较长的寿命、良好的灵活性和机械稳定性,表明Fe-NSDC催化剂在可充电和便携式设备中具有极大的应用潜力[图14(D)和(E)].
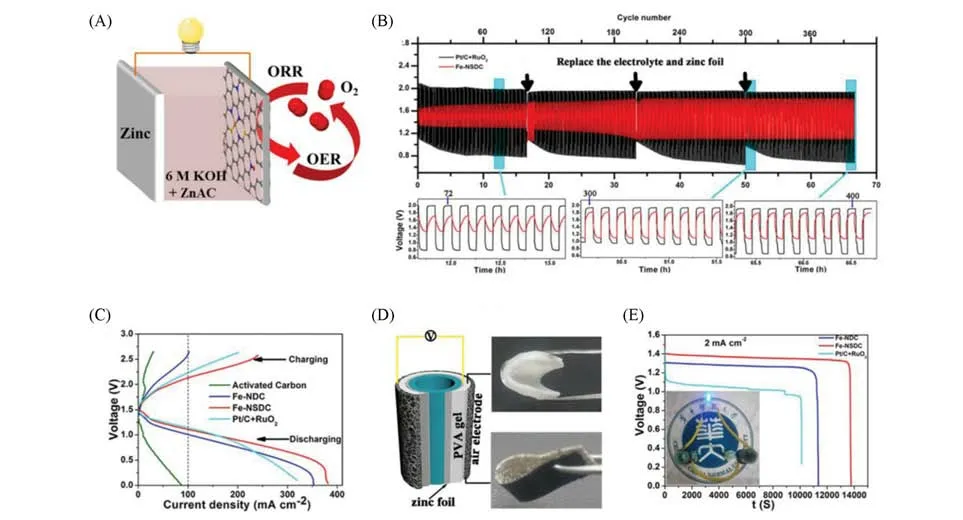
Fig.14 Schematic of the rechargeable Zn‐air battery(A),galvanostatic discharge‐charge cycling curves using Pt/C+RuO2 and Fe‐NSDC catalysts at 4 mA/cm2 with 10 min cycle(B),charge and discharge polarization curves of rechargeable Zn‐air batteries using different catalysts as air electrode(C),schematic diagram of the flexible all‐solid Zn‐air battery(D),the galvanostatic discharge curves of a coiled all‐solid Zn‐air battery at current density of 2 mA/cm2 and a corresponding image of two batteries in series to power a blue LED light(E)[84]
本课题组也在单原子催化剂用于可充放电固态锌-空气电池的方面取得了一些进展,Zhao等[87]开发了一种双层ZIF 的策略,将含有乙酰丙酮铁的ZIF-8 外延生长在所制备的ZIF-8 核之外(ZIF-8@Fe/ZIF-8),成功制备了一种负载在逐级多孔CN 载体上的富缺陷的单原子Fe 催化剂(Fe1/d-CN).以Fe1/d-CN催化剂作为空气阴极组装的柔性准固态锌-空气电池在碱性和中性电解质中均表现出优异的性能,开路电压分别为1.50和1.20 V,最大峰值功率密度分别为78.0和15.8 mV/cm2,并具有良好的充放电耐久性和机械柔韧性[图15(A)].
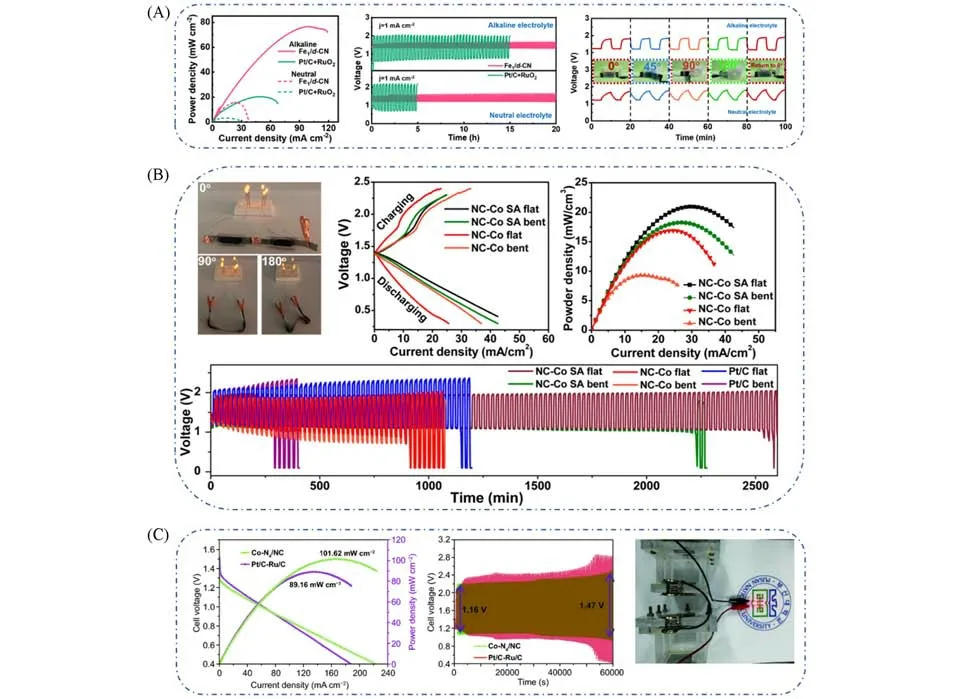
Fig.15 Power density curves of flexible quasi‐solid‐state alkaline(solid line) and neutral(dotted line)Zn‐air batteries,comparison of stability test between the Fe1/d‐CN and Pt/C+RuO2 catalysts as the air ca‐thode in flexible quasi‐solid‐state alkaline(top side) and neutral(bottom side)Zn‐air batteries,cycling stability of the quasi‐solid‐state Zn‐air battery in alkaline(top side)and neutral(bottom side) electrolytes with different bending angles(A) [87],photograph of 6 LEDs powered by two assembled Zn‐air batteries with different bending angles(0°,90°,180°),discharge and charge polarization curves,power‐current density curves,comparison of the cycling stabilities in both bent and flat states(B)[88],discharging polarization and power density curves based on the Co‐N4/NC and Pt/C‐Ru/C catalyst,cycling test(100 cycles) at a current density of 10 mA/cm2 with Co‐N4/NC and Pt/C‐Ru/C catalyst,images of two home‐made rechargeable Zn‐air batteries in series with a LED(C)[89]
Zang等[88]以MOF材料为前驱体,采用简易碳化-酸洗工艺制备了一种组装在氮掺杂多孔碳纳米片阵列上的Co单原子催化剂(NC-CoSA).与Co纳米颗粒(NC-Co)相比,NC-CoSA催化剂具有显著增强的OER 和ORR 活性,OER 过电位和ORR 半波电位分别为360 mV 和0.87 V.如图15(B)所示,以NC-CoSA 催化剂组装的柔性准固态锌-空气电池具有良好的机械灵活性和稳定性,在充放电测试曲线中,在电流密度达到10 mA/cm2时,NC-CoSA催化剂的电压间隙远小于NC-Co催化剂,表明其具有更优异的充放电能力.此外,NC-CoSA 基柔性准固态锌-空气电池在其平坦状态下的最大电流密度为31.0 mA/cm2,峰值功率密度为20.9 mW/cm3,也优于NC-Co(24.7 mA/cm2,16.9 mW/cm3).最后,比较了NC-CoSA,NC-Co和Pt/C催化剂作为空气电极的柔性固态锌-空气电池在平坦和弯曲状态下的长期循环稳定性,观察到开发的NC-CoSA基柔性准固态锌-空气电池具有更优异的稳定性,表明NC-CoSA催化剂在可充电锌-空气电池中具有巨大的应用潜力.
Chen 等[89]通过超声等离子工程制备了Fe-N4/NC 和Co-N4/NC 单原子催化剂,实验中以高纯度苯胺作为溶剂和NC 基体混合作为前驱体,Fe/Co 金属酞菁作为偶联剂,经超声等离子工程处理获得具有Fe/Co-N4结构的单原子催化剂.Co-N4/NC的OER和ORR双功能电位间隙仅为0.79 V,优于Pt/C-Ru/C催化剂,理论计算结果表明,Co-N4是O2吸附-解吸附机制的主要活性位点.如图15(C)所示,在实际的液态锌-空气电池测试中,Co-N4/NC 基空气阴极电池表现出突出的容量(762.8 mA·h·g−1)和功率密度(101.62 mW/cm2),超过了Pt/C-Ru/C催化剂作为空气阴极的电池(700.8 mA·h·g−1)和89.16 mW/cm2).此外,Co-N4/NC基液态锌-空气电池在100次充放电循环后,电位偏差从1.16 V仅增加到1.47 V,表明其具有良好的充放电稳定性.
5.2 双功能HER-OER催化剂的全解水性能
由于全解水可以生产清洁环保的氢能源,且没有二氧化碳的产生,这种制氢方法被认为是一种潜在的解决能源和环境问题的策略[90,91].全解水装置涉及HER和OER两个半反应,其利用可再生电能制备的绿色氢能作为原料转化产生电能,用作日常生产生活.因此,该装置为清洁可再生能源的转化和储存提供了一个有前途且环保的途径[92~94].然而,在实践中HER和OER存在动力学迟缓的缺点,反应的顺利进行需要更高的电势驱动,单原子催化剂的快速发展有望解决这一问题.
单原子催化剂可以通过最大化的原子利用率降低成本,但由于其自身的高表面能状态易诱发团聚,通常会使性能衰减,尤其在严苛的酸性条件下保持其优异的性能极为困难.单原子Ir催化剂的稳定性问题使其尚未应用于酸性OER领域,为了解决单原子Ir催化剂在酸性OER中的稳定性问题,Luo等[95]构思并实现了一种双重保护策略,通过将Ir原子分散在Fe纳米粒子上,并将IrFe纳米粒子嵌入氮掺杂碳纳米管中得到Ir-SA@Fe@NCNT催化剂.在仅1.14 μg/cm2的低负载下,Ir-SA@Fe@NCNT催化剂表现出优异的OER和HER活性,在0.5 mol/L H2SO4的酸性条件下,在10 mA/cm2电流密度下的OER和HER 过电位分别为250 和26 mV.鉴于此,从图16(A)的全解水性能测试中可以观察到,在酸性条件下,仅需要1.51 V 的电位就可以达到10 mA/cm2的电流密度来驱动水分解.此外,如图16(B)~(D)所示,Ir-SA@Fe@NCNT 催化剂在10 和100 mA/cm2的电流密度下均具有比Pt/C-IrO2催化剂更杰出的稳定性.Dou 等[60]利用ZIF-67 作为前驱体合成了一系列单原子催化剂,其中Ir1@Co/NC 具有最优异的OER和HER活性,全解水装置在实现10 mA/cm2的电流密度时的电位仅为1.603 V,展现出优异的全解水性能[图16(E)和(F)].
Lee 等[66]开发的CoSAs-MoS2/TiN NRs 催化剂,在全pH 条件下具有优异的OER 和HER 活性.为了评估在实际应用中使用CoSAs-MoS2/TiN NRs 催化剂作为电极材料的可能性,在全pH 条件下采用该催化剂作为全解水装置的阴极和阳极,并对其性能进行了分析.如图17(A)~(I)所示,CoSAs-MoS2/TiN NRs 电极在酸性、碱性和中性电解质中,在10和50 mA/cm2的电流密度时,其工作电压分别为1.70和2.00 V,1.65和2.02 V,1.66和2.26 V,具有可与贵金属Pt/C//RuO2催化剂相当的全解水活性.通过计时电流测试评估了CoSAs-MoS2/TiN NRs催化剂的全解水稳定性,在酸性、碱性和中性电解质中进行了30 h 的稳定测试,发现CoSAs-MoS2/TiN NRs 催化剂分别保留了原始电流密度的96.9%,98.5%和93.14%.此外,经30 h 计时电流测试后的极化曲线也基本没有明显的变化,证明了CoSAs-MoS2/TiN NRs催化剂在全pH条件下的全解水过程中均具有良好的稳定性.
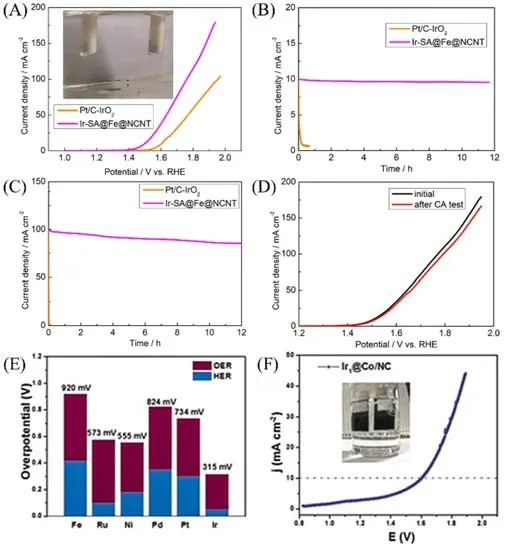
Fig.16 LSV curves of water splitting test in 0.5 mol/L H2SO4 electrolyte of Ir‐SA@Fe@NCNT and Pt/C‐IrO2(A),chronoamperometric tests of Ir‐SA@Fe@NCNT and Pt/C‐IrO2 for achieving 10 mA/cm2(B) and 100 mA/cm2(C),LSV curves of water splitting test in 0.5 mol/L H2SO4 electrolyte of Ir‐SA@Fe@NCNT before(black line)and after(red line)chronoamperometric test(D)[95],the overall overpotential of the corresponding electrodes obtained at 10 mA/cm2(E),the polarization curves of overall water splitting by the Ir1@Co/NC catalyst(F)[60]

Fig.17 Overall water‐splitting measurements with a symmetrical electrode system using CoSAs‐MoS2/TiN NRs as both cathode and anode in 0.5 mol/L H2SO4(A),1.0 mol/L KOH(B)and in 1.0 mol/L PBS(C),in comparison with the Pt/C//RuO2 couple,a comparison of operating voltages at J values of 10 and 50 mA/cm2 between CoSAs‐MoS2/TiNNRs and Pt/C//RuO2 in 0.5 mol/L H2SO4(D),1.0 mol/L KOH(E)and in 1.0 mol/L PBS(F),chronoamperometric curves of the CoSAs‐MoS2/TiN NRs in 0.5 mol/L H2SO4(G),1.0 mol/L KOH(H)and in 1.0 mol/L PBS(I)[66]
Li 等[96]通过自牺牲策略制备了同时集成有Co 单原子与Co9S8纳米粒子的中空碳纳米管催化剂(CoSA+Co9S8/HCNT).其独特的结构使CoSA+Co9S8/HCNT催化剂能够作为ORR,OER和HER的高效三功能电催化剂.CoSA+Co9S8/HCNT 催化剂显示出良好的ORR 和OER 活性,Ej=10和E1/2之间的电位差(ΔE)仅为0.705 V,远小于Pt+RuO2的0.777 V.此外,CoSA+Co9S8/HCNT 催化剂对HER 也具有较高的活性,在10 mA/cm2时HER 过电位为250 mV,接近商业Pt/C 催化剂.由于CoSA+Co9S8/HCNT 催化剂对ORR,OER和HER具有杰出的三功能催化活性,将其应用于可充电锌-空气电池和全解水装置,研究其实际应用潜力.如图18(A)所示,所组装的锌-空气电池作为电源驱动组装的全解水装置,发现只需施加1.59 V的工作电压就可以达到10 mA/cm2的电流密度,远低于Pt/C+RuO2的1.66 V.该装置经过连续10 h的全解水后,电流密度仍保持了94.5%,表明CoSA+Co9S8/HCNT作为一种高效的多功能电催化剂,具有优越的耐久性.
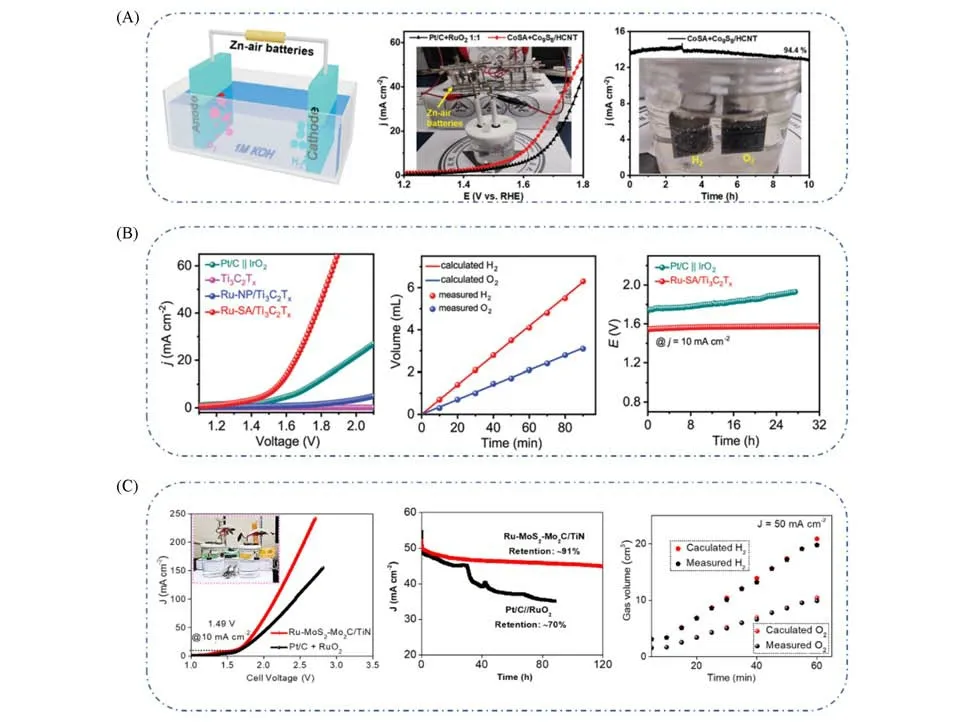
Fig.18 Schematic diagram of self‐power water splitting electrolyzer,LSV curve of the CoSA+Co9S8/HCNT electrodes(A)[96],geometric‐area‐normalized OWS LSV curves without iR compensation,the amount of generated H2 and O2 as a function of the reaction time at j=10 mA/cm2,stability test of Ru‐SA/Ti3C2Tx and Pt/C||IrO2 for OWS at j=10 mA/cm2(B)[97],LSV measurements of the Ru‐MoS2‐Mo2C/TiN and Pt‐C+RuO2‐based devices for overall water splitting in 1.0 mol/L KOH medium,stability of the Ru‐MoS2‐Mo2C/TiN and Pt‐C//RuO2‐based devices measured at an initial current response of 50 mA/cm2,faradaic efficiencies of the Ru‐MoS2‐Mo2C/TiN‐based device for water splitting(C)[98]
Peng 等[97]报道了一种具有高活性和耐久性的三功能电催化剂(Ru-SA/Ti3C2Tx),在超薄Ti3C2TxMXene 纳米片上负载原子分散的Ru-O2位点且Ru 的负载量(质量分数)达到0.23%.研究发现,Ru-SA/Ti3C2Tx催化剂在酸性条件下对HER,ORR 和OER 具有极高活性和稳定性.值得注意的是,当Ru-SA/Ti3C2Tx作为酸性全解水装置的阳极和阴极时,在1.56 V的工作电压下即可达到10 mA/cm2的电流密度,表明其在全解水装置中良好的应用潜力[图18(B)].此外,收集了阴极和阳极上的气体产物,检测到H2和O2的摩尔比接近2∶1.最重要的是,Ru-SA/Ti3C2Tx基全解水装置在32 h的稳定性测试中性能没有明显衰减,表明其在酸性条件下具有优异的稳定性.
Hoa 等[98]报道了一种负载单Ru 原子的MoS2-Mo2C 异质结构纳米片,其覆盖在高导电性的一维氮化钛纳米棒阵列上,形成独特的三维分层多孔材料(Ru-MoS2-Mo2C/TiN).Ru-MoS2-Mo2C/TiN 催化剂在1 mol/L KOH 介质中表现出优异的HER 和OER 活性,在10 mA/cm2电流密度下,过电位分别为25 和280 mV.如图18(C)所示,由Ru-MoS2-Mo2C/TiN 组装的全解水装置在10 mA/cm2电流密度时所需的工作电压仅为1.49 V,低于同条件下的Pt/C+RuO2.在稳定性测试中发现,基于Ru-MoS2-Mo2C/TiN 的全解水装置在120 h 测试后性能仅衰减约9%,而基于Pt/C+RuO2的全解水装置在较短的运行时间后就达到30%的性能衰减,表明Ru-MoS2-Mo2C/TiN 基全解水装置比Pt/C+RuO2基全解水装置更稳定.此外,在研究该装置将电能转换为H2和O2的效率后发现,Ru-MoS2-Mo2C/TiN 基全解水装置产生H2和O2的法拉第效率分别为97.2%和96.1%,表明大部分提供的电能用在全解水过程中产生H2和O2.
5.3 其它类型双功能单原子催化剂的应用
最近,甲醇[99]、脲[100]、5-羟甲基糠醛[101]、苯甲醇[102]和肼氧化反应[103,104]等可以替代氧气析出反应作为阳极替代反应的研究被相继报道,以实现超低电池电压下全解水或生产其它增值产品.Li等[74]合成了一种WS2/Ru SAs 催化剂,将其作为阳极和阴极的催化材料,在10 mA/cm2时,所需电压仅为15.4 mV,远低于包括商用Pt/C在内的大多数催化剂.如图19(A)所示,肼氧化反应辅助全解水的数码照片可以清楚地观察到氮气和氢气的剧烈析出.Gan等[105]利用制备的Fe-Nx-CSACs催化剂作为电解装置的阴极和阳极,实现了对阴极EER和阳极ACR高效的催化转化,建立了以Fe-Nx-CSACs作为阴极和阳极的电化学EER||ACR 的耦合系统[图19(B)].在该系统中,Fe-Nx-CSACs 催化剂的性能高于商业20% Pt/C.此外,对氯苯甲醚(313.3~329.6 μmol·mg−1·h−1)、邻氯苯甲醚(39.6~45.7 μmol·mg−1·h−1)、乙烯(220.2~250.3 μmol·mg−1·h−1)和氯乙烯(6.7~9.5 μmol·mg−1·h−1)的产率及对氯苯甲醚(46.5%~55.0%)和乙烯(38.3%~43.7%)的法拉第效率在经过6 个连续的电解循环后仍得以保持,表明该耦合系统具有良好的可重复性.
可充电金属-CO2电池结合了CO2RR和金属-空气电池的工作原理,作为一种新型可持续能源转换和存储技术,有利于促进碳循环利用.而电池运行过程中产生的Li2CO3和Na2CO3的高分解能垒严重损害了Li-CO2和Na-CO2电池的可逆性.因此,具有高效双功能活性的单原子催化剂被认为是解决该难题的阳极催化剂候选材料之一.Hu等[106]通过将单分散的Fe原子植到入3D多孔碳中,合成了一种用于可充电Li-CO2电池中CO2RR和二氧化碳析出反应(CO2ER)的高效催化剂(Fe-ISA/N,S-HG),不同于传统Li-CO2电池,放电产物(Li2CO3)在充电时很容易分解.所得的可充电Li-CO2电池在100 mA/cm2时表现出约1.17 V 的低电位间隙,并且可以在高电流密度下(1 A/g)可重复充放电超过200 次,容量保持为1000 mA·h·g−1.
Zeng 等[79]制备了一种NiFe 双金属单原子催化剂,其具有优异的CO2RR 和OER 催化活性,将其作为Li-CO2电池阴极,锌板为阳极组装成可充电的Zn-CO2电池.当电流密度为0.1~10 mA/cm2时,放电(充电)电压位于0.89~0.12 V(2.16~3.01 V)范围内,表明此可充电Zn-CO2电池的实用性.如图19(C)所示,所组装的电池在放电电流密度为5 mA/cm2和充电电流密度为2 mA/cm2下,恒定电流放电-充电循环测试显示,其在180次循环期间可以提供稳定的电压输出.

Fig.19 Schematic illustration of the HzOR‐assisted OWS mechanism in two‐electrode H‐type electrolyze using CC@WS2/Ru‐450 as both anode and cathode(A)[74],optical image of the two‐electrode configu‐ration schematic illustration for EER||ACR(B)[105],galvanostatic discharge‐charge cycling curves of rechargeable Zn‐CO2 battery cell at the discharge current density of 5 mA/cm2 and charge current density of 2 mA/cm2 for 180 cycles(C)[79]
6 总结与展望
本文总结了具有双功能活性的单原子催化剂的最新研究及其在电化学应用中的进展.随着单原子催化剂的迅速发展,具有双功能活性的单原子催化剂因其可显著拓展催化剂的实际应用范围而受到广泛关注.这不仅与单原子催化剂自身的低配位环境、充分暴露的活性中心、高催化活性与选择性、高原子利用率等优势密不可分,而且与社会对于加快可持续能源器件发展的需求相符合.本文首先介绍了单原子催化剂的发展历史以及独特优势,然后介绍了各类具有双功能活性的单原子催化剂的最新研究进展,最后介绍了具有双功能活性的单原子催化剂在可持续能源器件中的应用研究,其中包括受到广泛关注的金属-空气电池及全解水装置.
尽管各类具有双功能活性的单原子催化剂已经被广泛开发,并具有优异的性能,但仍存在一些问题亟需解决:首先,由于单个原子的极大表面能,保持催化剂的稳定性仍存在很大的挑战;其次,单一功能的单原子催化剂的设计合成已经被大量报道,而因同一催化剂中双功能活性之间的平衡难以协调,使得具有双功能活性的单原子催化剂的设计和制备仍面临挑战;最后,具备双功能活性的单原子催化剂对相应的双功能活性的协同催化机理仍不清晰,需要进一步探索.值得注意的是,现有的研究已经更多集中于非贵金属单原子催化剂:一方面是为了满足催化剂降低自身成本的需要;另一方面对于当今社会的可持续发展具有重要意义.
综上所述,在未来,更加廉价、高效、易合成的具有双功能活性的单原子催化剂的设计合成值得深入研究,尤其要重视发展可以实现清洁化生产的、高效绿色的非贵金属单原子催化剂.因此需对催化剂的双功能性机理进行深入分析,以提供对材料设计制备的指导,探索出一种普适性更强的策略,以推动具有双功能活性的单原子催化剂的快速发展及其在可持续能源器件中的实际应用.
