单原子催化材料在电化学氢循环应用中的研究进展
2022-06-29陈长利米万良李煜璟
陈长利,米万良,李煜璟
(1.北京理工大学材料学院,北京 100081;2.中国石油化工股份有限公司石油化工科学研究院,北京 102299)
近年来,日益增长的能源需求和环境危机促使了可替代的高效能源转换和储存技术的广泛研究.氢能被认为是高能量密度、零污染的清洁能源,通过氢和水之间的相互转化可以实现一个能源循环的闭环.通常氢能的利用包括制氢、储氢、运氢和用氢4个环节,其中制氢和用氢是核心环节.电解水制氢技术可高效地将电能转化成氢能,实现按需供给的分布式氢能源的推广应用,其优势在于制得的氢气纯度高,对氢气的高效利用具有重要意义.氢燃料电池是将氢分子中储存的化学能直接转化成电能的电化学装置,涉及到的化学反应和电解水为逆过程,整个反应过程效率高、无污染.电化学催化则是电解水和燃料电池中的核心化学过程,决定能量的转化效率和速率.因此,目前关于电解水制氢和燃料电池的基础研究主要集中在高效催化材料的设计方面.
目前,商用电催化材料普遍使用负载型贵金属纳米粒子,通过降低活性纳米颗粒尺寸,从而提升贵金属使用效率并降低其载量,是该领域持续研究的热点[1,2].当尺度缩小到单原子尺度,金属原子的使用效率达到理论上的最大化.Zhang等[3]于2011年报道了首个Pt单原子催化材料(Pt1/FeOx),并将其应用于CO氧化反应中,自此引发了人们对单原子催化的研究兴趣.单原子催化材料因其高效的原子利用率和特殊的催化性能而受到广泛关注,其特殊的化学结构使其在不同催化反应中表现出独特的催化活性和选择性,构建高效原子尺度催化材料成为近年来新兴的研究热点[4,5].单原子催化剂(Singleatom-catalysts,SACs)作为均相和非均相催化材料之间的桥梁,满足了对负载金属高效化利用的迫切需求,也突破了材料设计方法上的瓶颈.对电催化性质和结构-活性关系的深入理解,启发了学术界在原子水平上准确调整催化活性位点的相关研究.此外,催化活性位点的不均匀性和它们之间复杂的相互作用对基础研究提出了严峻的挑战.如图1 所示,近10年来,单原子催化材料的开发和应用得到迅速发展,特别是在化学、物理、材料科学及工程等领域.其中,单原子催化材料在电化学制氢和氢燃料电池领域的开发和应用被认为是最具有前景的研究方向[6,7].如图1 中的插图所示,单原子催化材料在电化学氢循环所涉及的研究约占近10 年全部单原子催化材料研究的1/5,其中,单原子催化材料在氧还原反应(ORR)、析氢反应(HER)以及析氧反应(OER)催化的研究中成为热点.
Pt基纳米颗粒被认为是最高效的析氢反应(Hydrogen evolution reaction,HER)催化材料.具有高原子利用率的Pt单原子催化材料(Pt SACs),有望解决因低储量和高成本使Pt基催化材料不能进一步应用的问题.Sun等[2]采用原子层沉积技术(Atomic layer deposition,ALD)制备了Pt SACs并将其用于催化HER,与商业催化材料Pt 相比,Pt SACs 催化活性提高了37 倍且催化稳定性有显著提升.Chen 等[8]采用热解-蒸发的策略设计合成了单原子Fe-N4催化剂,该单原子催化材料表现出优异的全解水活性和优异的催化稳定性.计算结果表明,在Fe-N4位点上的析氧反应(Oxygen evolution reaction,OER)过电势显著降低,同时Fe-N4位点可以显著降低H*吸附能(ΔGH*).Li等[9]通过理论计算研究了一系列负载在磷钼酸(PMA)团簇上的金属单原子催化材料的电催化活性.通过PMA稳定的金属单原子具有较高的稳定性和催化活性,其中Pt1PMA,Ru1PMA,V1PMA 和Ti1PMA 的ΔGH*=0,表现出优异的HER 催化性能;而Co1PMA 和Pt1PMA 表现出较好的OER 催化活性.Chou 等[10]对比研究了Ir,Pt,Ru,Pd,Fe和Ni 等金属单原子催化材料,发现Ir SACs 表现出最优异的电解水制氢活性,当电流密度达到10 mA/cm2时,仅需施加1.65 V的电位,在催化循环5 h之后仍保持稳定的催化活性.Shui等[11]发现单原子催化剂Ir1-N/C在质子交换膜燃料电池(Proton exchange membrane fuel cells,PEMFCs)测试中表现出良好的催化活性,峰值功率密度达到了870 mW/cm2(H2-O2),是商业Ir/C 催化材料的2 倍.同时,测试结果表明,Ir1-N/C在PEMFCs 测试中具有良好的稳定性.Xing 等[12]报道了一种Ir 纳米颗粒(NPs)和Ir SACs 协同作用的IrNP@IrSA-N-C催化剂,用于解决PEMFCs中催化材料CO中毒的问题.研究表明,Ir单原子不仅可以作为CO的氧化活性位点,同时可以清除掉吸附在Ir纳米颗粒上的CO,从而使得Ir纳米颗粒上有充足的活性位点用于H2的氧化反应(Hydrogen oxidation reaction,HOR).Wu等[13]基于Co基金属有机骨架(Metalorganic frameworks,MOFs)合成的Co 单原子催化剂20Co-NC-1100 具有优异的PEMFC 测试活性和稳定性.在H2-O2和H2-air测试环境中的峰值功率密度分别为0.56 和0.28 W/cm2,且可以保持长达100 h的测试稳定性.
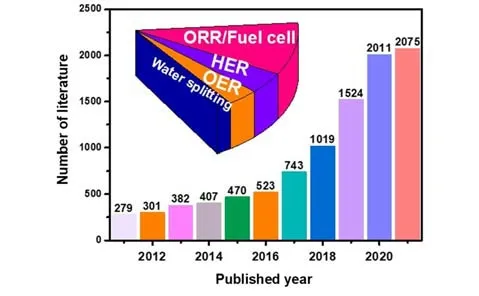
Fig.1 Numbers of published literatures from 2011 to 2021 by the keyword“single atom catalysts”in all data⁃base of web of science
影响单原子催化材料的活性、稳定性以及选择性的因素主要包括单原子金属的种类、单原子金属与基底间的相互作用以及单原子金属与临近团簇或者纳米颗粒的协同作用.了解单原子催化材料的构效关系,对开发高效稳定的催化材料具有重要意义.本文综合评述了近年来SACs的设计(高效性、稳定性以及选择性设计)以及SACs在电化学氢循环(电解水制氢、氢燃料电池)领域的应用进展,并对其面临的挑战和发展前景进行了展望.
1 单原子催化材料的设计
1.1 单原子催化材料的结构设计
单原子催化材料的优化主要集中于调控活性位点结构,通过微观环境对金属活性位点进行调节,提高单原子催化材料的活性和稳定性[14~16].研究表明,微观调控不仅可以调节金属位点的电子结构、改善优化催化活性位点的空间结构,同时邻近原子可以作为活性位点参与相应的催化反应,最终达到增强催化活性和稳定性的目的[17~19].基于此,可以将固定金属活性位点的方法分为杂原子(N,S,P,B)配位效应[20]、空位效应[21]、掺杂策略[22]以及离子交换策略[23].
1.1.1 配位效应 金属原子在碳载体上通过与杂原子配位构成配位环境可调的单原子催化结构,是目前单原子催化材料设计的主要手段.该方法主要通过杂原子与金属原子中的电子空轨道进行配位实现电子结构的调控[20,24,25].Zhang 等[26]通过杂原子配位锚定的方法将金属原子固定在S,N共掺杂的碳材料上,得到了具有不同配位结构的单原子催化材料.如图2(A)所示,以SiO2纳米球作为软模板,将不同金属(Fe,Co,Ni)前驱体和烯丙基硫脲在80 ℃下混合,烘干热解得到金属单原子催化材料.研究表明,金属的种类不同,得到的活性位点结构不同,因此,可以通过选择不同的金属和杂原子进行组合,实现特定需求的单原子活性位点的设计.
1.1.2 空位效应 通过空位效应将金属单原子锚定在载体上,是构建稳定高效单原子催化材料的一种有效手段[21,27].即,通过一定的方法制造原子空位作为金属单原子的吸附位点,随后将目标单原子锚定在含有空位的载体上,形成稳定高效的单原子催化材料.Yang等[28]制备了富有缺陷石墨烯用于锚定单原子Pt,最终形成具有Pt-C3构型的催化活性位点.如图2(B)所示,将石墨烯和三聚氰胺混合之后,高温热解得到N掺杂石墨烯,随后升温至N原子完全去除,最后通过光还原法将Pt固定在缺陷处形成Pt 单原子催化材料.Zheng 等[29]采用空位锚定策略合成了一系列负载在过渡金属硫化物(Transitional metal dichalcogenides,TMCs)上的单原子催化材料,该方法首先对TMCs进行剥离制造S空位,这些S空位为硫脲-过渡金属配合物提供了配位点,热处理之后获得了高密度的单原子催化材料.
1.1.3 掺杂策略 掺杂策略主要是将特定金属单原子掺入到某些金属氧化物、磷化物和硫化物等晶体结构中,该方法不仅可以保证催化材料结构的稳定性,同时还可以提高催化材料的活性和稳定性.Wang等[22]采用自上而下的磷化策略制备了单原子Ir掺杂的Ni2P催化材料,该策略包括两步:首先是油相高温将Ir和Ni的前驱体还原为Ir-Ni纳米颗粒,然后加入三苯基磷进行高温磷化.Chen等[30]将W原子以取代Mo位点的方式掺入到MoP的晶格之中,成功制备了W单原子掺杂的MoP催化剂W0.25Mo0.75P/PNC.如图2(C)所示,将W,Mo金属前驱体与三聚氰胺、石墨烯以及磷酸在室温下初步混合,得到均匀的悬浊液,烘干之后高温热处理,得到单原子催化材料.
1.1.4 离子交换策略 离子交换策略是基于一部分金属原子(如Zn)在高温处理过程中易被蒸发掉,而作为活性位点的金属被吸附在留下的金属空位处,最终形成目标单原子催化材料[23].Xiao等[31]采用基于MOF 空间限制和离子交换策略制备了Ru单原子催化材料Ru SAs-NC.如图2(D)所示,沸石咪唑酯骨架结构(ZIF-8)的空腔直径为1.16 nm,可以作为分子尺度的活性位点边界,封装直径约为1.04 nm的金属前驱体Ru(acac)3分子,为Ru原子的聚集提供防护栅栏.随后,Zn原子在高温处理中蒸发,留下的富N缺陷为Ru3+提供了大量的吸附位点,最终形成Ru单原子催化材料.Shan等[32]通过MOF介导的离子交换-热解过程,将Ir成功地分配到尖晶石钴氧化物结构的阳离子亚晶格中.研究表明,Ir取代位点与尖晶石钴氧化物主晶格之间存在空间相关性,并且这些Ir单原子表现出短程有序特性.
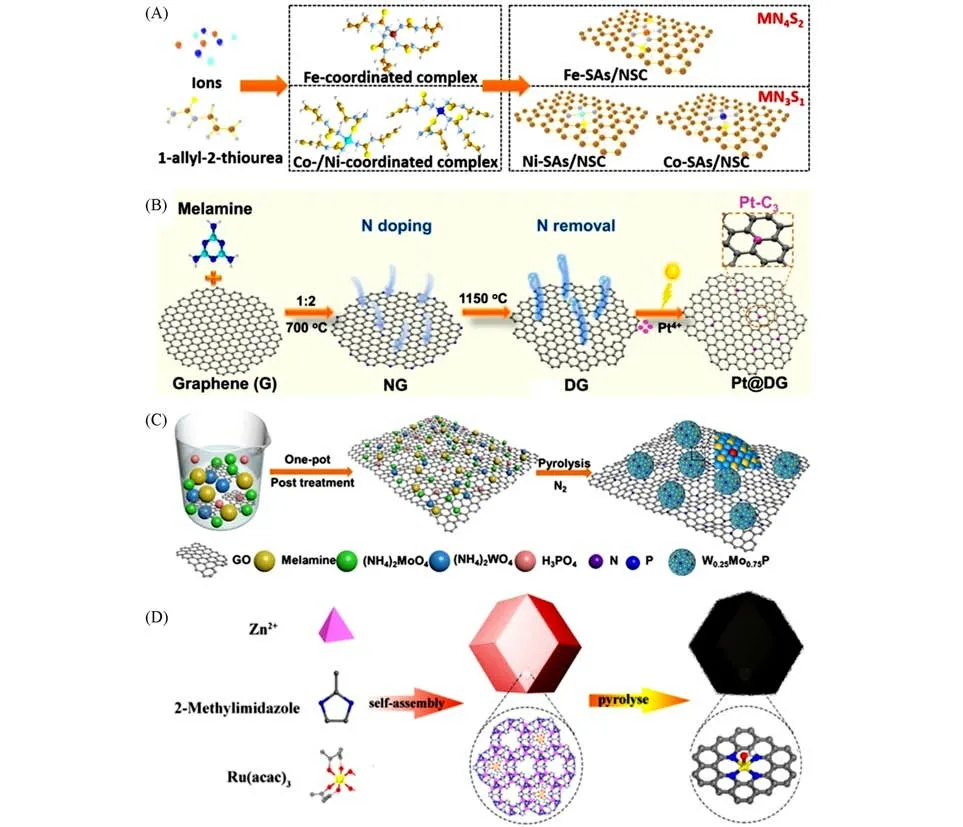
Fig.2 Preparation and morphology characterization of Fe⁃SAs/NSC(A)[26],schematic illustration of the prepara⁃tion of Pt@DG(B)[28],schematic illustration of the synthetic process for W0.25Mo0.75P/PNC(C)[30]and scheme showing host⁃guest strategy for the fabrication of single⁃atomic⁃site catalysts(D)[31]
1.2 单原子催化材料的功能化设计
1.2.1 选择性 单原子催化材料独特的化学结构是决定其催化选择性的关键[33].孤立的金属单原子位点无法为催化反应提供连续的催化活性位点,特别是那些需要多个相邻活性位点串联工作的反应,单原子催化剂只能选择性地催化生成中间产物[34].
研究发现,增加单原子金属的载量可以加强临近单原子之间的相互作用,从而实现多个催化位点的协同合作,最终完成多电子催化反应[35].Lee等[36]通过将金属Pt负载在氮化钛(TiN)上形成稳定的单原子Pt催化材料0.35%Pt/TiN(Pt质量分数).与Pt NPs 不同,合成的0.35%Pt/TiN 单原子催化材料在电化学氧还原反应(Oxygen reduction reaction,ORR)中高选择性地产生过氧化氢(H2O2),这是由于单原子Pt 位点改变了ORR 向二电子路径进行.同时,由于临近Pt 位点的缺失而不能为甲醇氧化提供必需的CO吸附位点,导致0.35%Pt/TiN 在甲醇电催化氧化过程中不具有催化活性.Tang等[37]通过对活性位点周围第一配位层和第二配位层更深层次的研究,发现配位环境的差别导致了催化过程的分化,可在单原子Co催化剂上实现四电子ORR向二电子ORR过程的转换.Zhang等[38]发现不同的金属单原子对催化反应具有高度的选择性,Pt1/PMA,Ru1/PMA,V1/PMA和Ti1/PMA表现出接近理想()的值,具有较好的HER催化活性,Co1/PMA和Pt1/PMA具有优异的OER的活性和选择性,而非贵金属Fe1/PMA SAC表现出高效的ORR电催化活性.
1.2.2 稳定性 单原子催化材料的稳定性主要体现在两个方面:(1)制备过程中的热稳定性;(2)催化过程中的反应稳定性.相比于金属团簇和纳米粒子,单个金属原子具有更高的表面能,这使得单个金属原子更趋向聚集,最终导致单原子催化材料不能长时间稳定存在[39,40].通常,稳定存在的单原子金属通过金属键或配位键被稳定在金属合金或者载体的表面,单原子金属和基体之间的化学键合作用不仅避免了单原子的团聚,还有效调控了单原子金属的电子结构,从而达到提高催化活性的同时保证稳定性的目的[41,42].
Shao等[43]提出了一种由Ce辅助优先形成单原子Fe位点的策略,基于CeO2对Fe原子的空间约束和较强的捕获能力,在热处理过程中可以有效地抑制孤立Fe的团聚.Fe原子与CeO2中的晶格氧结合保证了单原子Fe 的稳定存在,从而使得Fe 负载量高达4.6%(质量分数).Zhang 等[44]采用高温煅烧的方法,通过共价键将金属Pt和基底Fe2O3稳定地键合在一起,达到了稳定Pt单原子的目的,最终制备了高载量、热稳定性良好的Pt 基单原子催化剂Pt1/Fe2O3.将单原子金属掺杂入金属氧化物中,实现非缺陷固定且高载量的单原子催化材料是一种很有应用前景的制备方法.可根据催化机制设计将不同的单原子金属和基底复合,实现催化目的.Zhang 等[38]研究了一系列多金属氧酸盐负载的Pt 催化剂的稳定性,以及Pt-载体相互作用对催化性能的影响,发现Pt原子更倾向于停留在多金属氧酸盐分子的4倍空穴位置,获得耐烧结Pt SACs.
2 电化学常用参数
近年来,单原子催化材料的研究在电化学领域发展迅速[45~49].下面将简单讨论单原子催化材料在电化学氢循环(电解水制氢和氢燃料电池)领域的应用.催化材料电催化性能好坏的确定需要建立有效的定量指标,即电化学描述符[50].
2.1 Tafel斜率
Tafel方程揭示了固-液界面电化学过程的反应机理和决速步骤(Rate determining step,RDS):

式中:η(V)为过电势;i(mA/cm2)为电流密度;a和b为塔菲尔常数,其中b为Tafel斜率.
如果在一系列的基元反应中有一个基元反应是RDS,则Tafel斜率可以反应RDS的特征[53].如HER反应,对于H吸附较弱的金属,其Tafel斜率接近120 mV/dec,反应的决速步骤为Tafel过程.而对H吸附较强的金属,由于存在吸附H的表面覆盖(如Pt上的欠电位沉积),导致反应过程复杂,其Tafel斜率减小(低于60 mV/dec),表明此时反应为Volmer-Tafel 机制[54].在实际研究工作中,由于催化材料的元素组成、表面粗糙度等不同,会降低Tafel斜率计算结果的准确性.此外,随着反应复杂程度以及电子转移数的增加,Tafel斜率的计算结果也会产生偏差.
2.2 转换频率
转换频率(Turnover frequency,TOF)通常用来评价催化剂的本征活性,该值用来衡量催化材料的催化反应速率,即单位时间内单个活性位点分子转化数[55].TOF的计算依赖于对活性位点或状态数量的准确测定,如下式所示,可以用来定量评价单原子催化材料的催化活性:

式中:I(A)为电流;NA(mol-1)为阿伏伽德罗常数;z为单个分子反应所转移的电子数;F(C/mol)为法拉第常数;ASD(m-2)为活性位点密度,即单位面积的活性位点的个数.
2.3 法拉第效率
法拉第效率(Faradic efficiency,FE)指实际生成物和理论生成物的百分比.在电化学催化中,可以理解为生成物量和消耗电量的百分比.在OER 和HER 催化过程中,通常需要用FE 评估催化剂的优劣.FE的值可以通过下式计算[30,56]:

式中:n为生成1 mol 目标产物转移的电子数;V为目标产物在气相色谱中检测出气体体积比;G(L/s)为气体体积流量;(A)为平均电流;F(96485 C/mol)为法拉第常数;P0(1.013×105Pa)为大气压力;R(8.314 J·mol-1·K-1)为理想气体常数;T0(K)为反应温度.
3 电解水制氢
电化学裂解水(电解水制氢)是一种很有前途的可再生产氢技术[57].如下式所示:
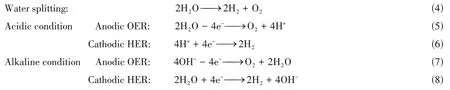
电解水包括两个半电池反应:在阳极上产生氧气OER[图3(A)]和在阴极上产生氢气的HER[图3(B)].半电池反应的催化效应共同主导了整体的电解水制氢效率,使得开发高效的HER和OER催化材料成为电解水制氢的关键.
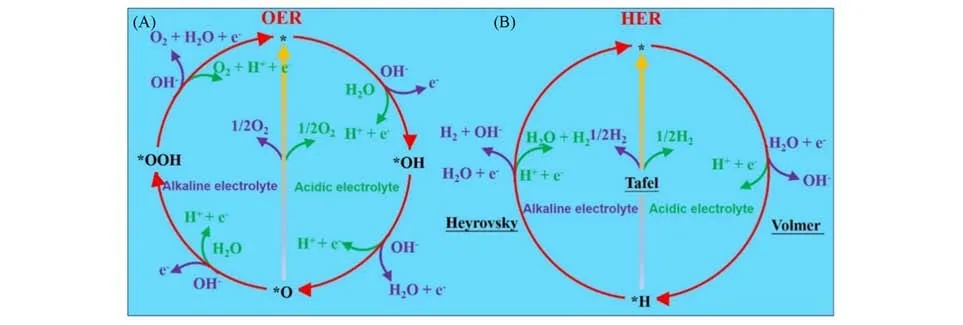
Fig.3 Elemental steps for the OER(A)and HER(B)in alkaline and acidic electrolytes[58]
3.1 析氢反应
HER作为电解水的阴极反应,如下式所示,其催化机制可以细分为3步[30,45,58]:
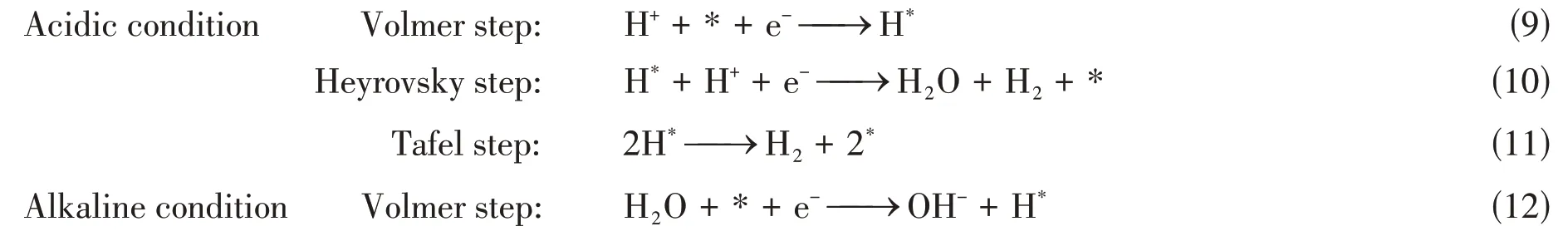
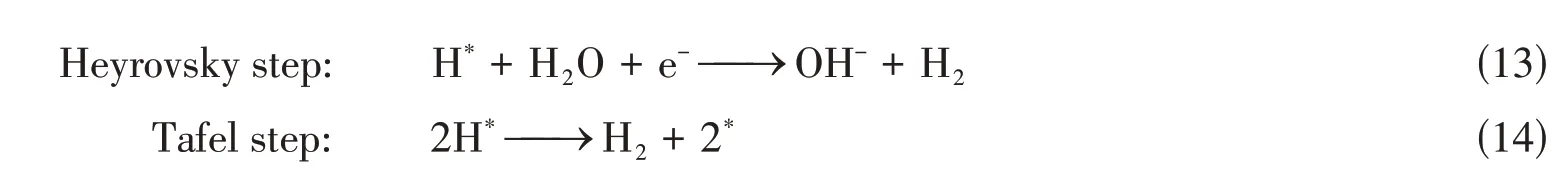
研究表明,在酸性介质中,H*吸附能(ΔGH*)的大小决定了HER 催化效率,而在碱性介质中,除ΔGH*外,水解离的动力学势垒(ΔGH2O)也决定整个反应的速率.无论是ΔGH*还是均在很大程度上取决于单原子催化材料的几何结构和电子结构,因此,单原子催化材料的合理设计是提高HER催化的核心[45,59].
Yang等[28]制备了具有Pt-C3活性位点的单原子催化剂Pt@DG,并将其应用于HER反应,其催化活性优于商业Pt/C催化材料,表现为在0.5 mol/L H2SO4和1 mol/L KOH 溶液中,HER过电位分别为30和37 mV.Zhou 等[60]通过电化学相变策略制备了一种锚定在二维Co(OH)2纳米片和Ag 纳米线(Ag NWs)复合结构上的单原子Pt催化剂[PtSA-Co(OH)2@Ag NW][图4(A)].如图4(B)所示,在1 mol/L KOH溶液中,PtSA-Co(OH)2@Ag NW表现出优异的HER催化活性,在10 mA/cm2电流密度处,其HER过电势低至29 mV,优于商业Pt/C 催化剂.部分状态密度(Partial density of states,PDOS)分析显示,Pt 原子与Co(OH)2轨道之间有较强的杂化,这些局域电子能级的增强有利于H2O的活化,进一步提高了HER的催化性能[图4(C)].此外,密度泛函理论(Density functional theory,DFT)计算表明,PtSA-Co(OH)2具有更低的ΔGH*(0.76 eV),表明金属-载体相互作用可进一步促进水的吸附和裂解动力学[图4(D)].Hossain等[59]利用DFT计算了N掺杂石墨烯上一系列过渡金属的催化活性,发现Co-SAC的催化活性最高,研究表明,Co-SAC中活性价态dz2轨道的位置接近于零,导致反键轨道部分填充,使其成为最活跃的HER轨道.
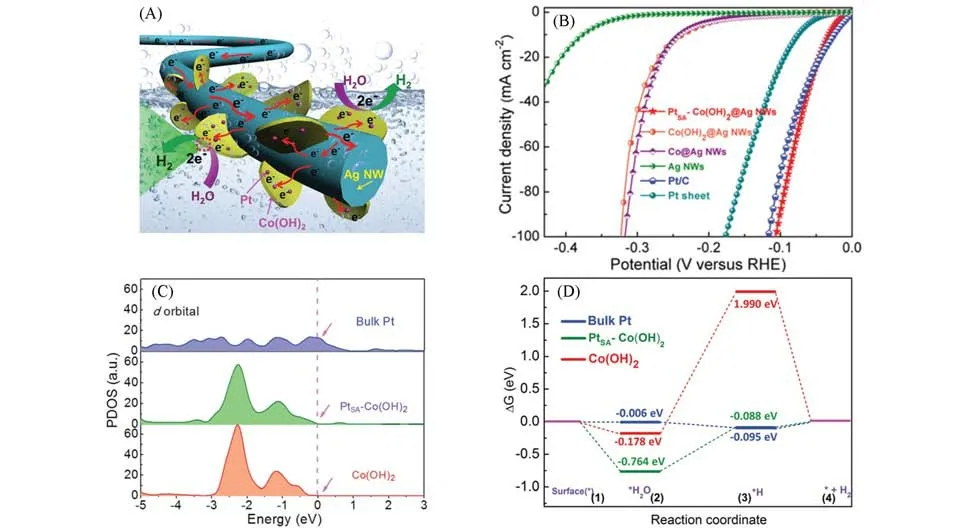
Fig.4 Seamlessly conductive electrode structure as a highly efficient electrocatalyst for the HER(A),linear sweep voltammetry(LSV) curves of PtSA⁃Co(OH)2@Ag NW,Co(OH)2@Ag NW,Co@Ag NW,Ag NWs,the Pt sheet and Pt/C(20%,mass fraction)(B),calculated PDOS of d orbitals of bulk Pt,PtSA⁃Co(OH)2 and Co(OH)2(C),calculated adsorption energies of H and H2O on the surface of Co(OH)2,PtSA⁃Co(OH)2 and bulk Pt(D)[60]
3.2 析氧反应
OER 因其缓慢的电化学动力学成为限制电解水制氢效率的关键反应,同时也是电池的氧化半反应[61].该过程涉及腐蚀电位下电子和质子的连续多步转移,导致过电位高,能量效率低,寿命有限[62~66].如下式所示,OER反应为直接四电子过程:
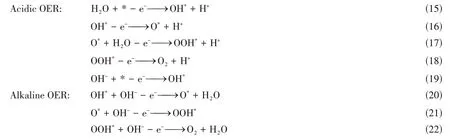
研究表明,OER催化活性可根据活性中间体的吸附自由能(ΔGOH*,ΔGO*,ΔGOOH*)来预测.碱性条件下,丰富的OH-以及较低的析氧活化能(ΔGOER)使得催化OER容易进行.此外,简易成熟的碱性电解水制氢装置也为碱性OER催化材料的开发提供了便利.Wang等[67]报道了一种超高负载的单原子Ir催化剂,并对该催化剂进行了OER催化性能研究,发现在碱性条件下,当电流密度到达10 mA/cm2时的过电位为215 mV.研究表明,负载的单原子Ir不仅是有利的催化活性位点,同时起到激活基体NiO表面活性的作用.Zhang等[68]将单原子Fe催化剂Fe1(OH)x/P-C用于加强OER催化中,结果表明,该催化剂的OER催化活性优于商业IrO2催化剂,在10 mA/cm2电流密度时的过电位低至320 mV,在0.35 V的过电势处,氧气的TOF 为0.62 s-1.理论计算结果表明,在Fe1(OH)x/P-C 上的OER 催化过程的决速步骤是由*O 生成*OOH 的O-O 偶联步骤.Fe1(OH)x/P-C 中的高价Fe4+中心提高了*O 中间体的亲电性,有利于OH-的亲核攻击形成*OOH,最终提高OER 催化活性.Li 等[69]采用NaCl 模板法制备了具有独特Ni-O-G(G代表石墨烯)结构的Ni单原子催化材料(Ni-O-G SACs),该催化材料表现出优异的OER活性和稳定性.如图5(A)所示,在碱性介质中,当电流密度达到10 mA/cm2时,Ni-O-G SACs的OER催化过电位为224 mV,远远低于商业RuO2催化材料.同时,该催化剂表现出优异的催化稳定性,在400 mV的高过电位下,显示出115 mA/cm2大电流密度,并保持50 h后无明显衰减[图5(B)].此外,在长期耐久性测试后,Ni-O-G SACs仍表现出均匀分散单原子分布,反映了Ni-O-G SACs 在OER催化过程中高度的结构稳定性.计算结果表明,Ni-O4(OH)2构型中Ni—O键的形成,高氧化态的Ni单原子降低了Ni-O-G SACs的吉布斯自由能和OER催化过电势,使得该催化材料在具有较高催化活性的同时也能保持优异的催化稳定性[图5(C)~(F)].
相较于碱性OER,酸性OER 面临的挑战主要来自于:(1)酸性条件要求更稳定的催化材料;(2)酸性条件不利于OH-吸附(热力学),表面OH-浓度低,使得表面电子参与的氧化过程更困难,且活化能更高(动力学),从热力学和动力学两个方面增加了OER转化过程的难度[70,71].
Yao等[72]通过压缩应变将原子分散的Ru固定到金属载体上(Ru1-Pt3Cu),促进了酸性OER过程,并减缓了Ru 基电催化剂在酸性电解质中的降解.在酸性OER 催化过程中,当电流密度达到10 mA/cm2时,催化过电位为220 mV,并且表现出比商业RuO2长10倍的使用寿命.基于氧化铱(IrOx)的材料是用于酸性介质中水电解最合适的OER催化材料.Zhang等[73]通过氧化IrAg合金将Ag单原子嵌入到IrO2基体中,制备了一种Ag1/IrOx单原子催化剂(Ag1/IrOxSAC).该催化剂在电流密度为10 mA/cm2时,OER过电位为224 mV,长期耐用性优于商用Ir(C-Ir).DFT 计算结果表明,Ag 原子的掺入显著降低了吸附自由能,促进了析氧过程.此外,较强的Ir—O 键防止了Ag1/IrOxSAC 中晶格氧的损失,从而提高了析氧反应的稳定性.Cao 等[74]报道了一种锚定在氮碳载体上的、具有Ru1-N4位点的单原子催化剂(Ru-N-C),并将其应用于酸性OER 催化中.在10 mA/cm2电流密度下,过电位为267 mV,质量活性高达3571 A/gmetal.此外,在长达30 h的耐久性测试中,该催化材料无明显的失活或分解现象,表现出了良好的OER催化稳定性.
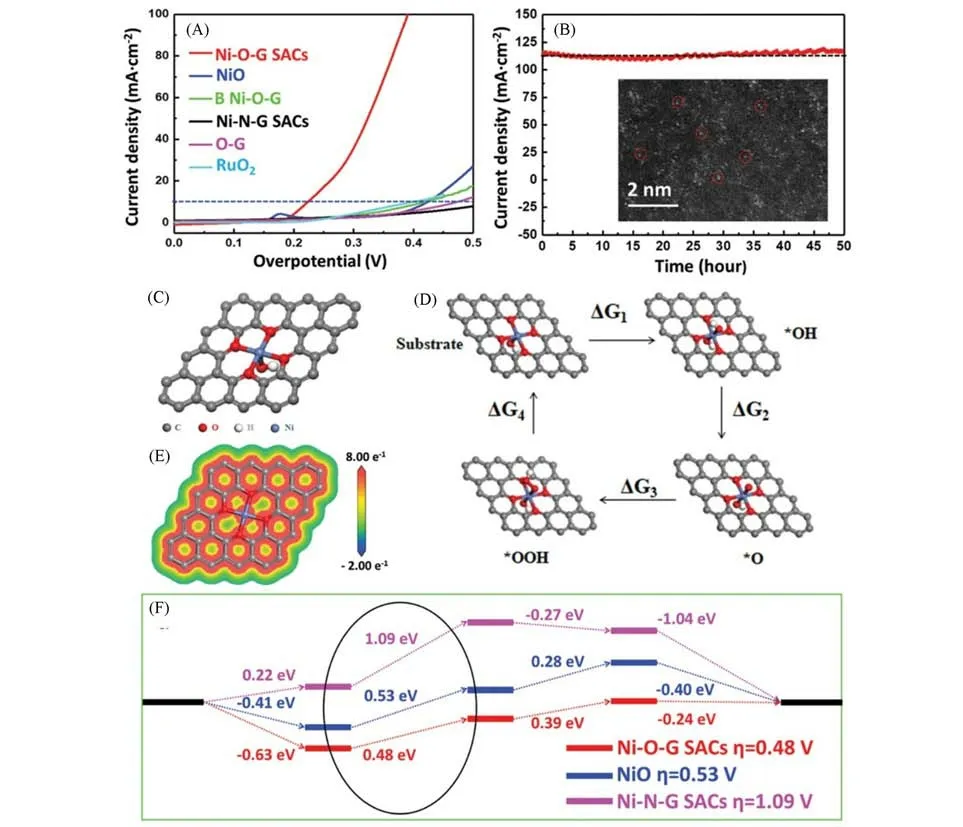
Fig.5 OER current curves of Ni⁃O⁃G SACs,NiO,B Ni⁃O⁃G,Ni⁃N⁃G SACs,O⁃G,and RuO2 in 1 mol/L KOH(A),chronoamperometric curve of Ni⁃O⁃G SACs obtained at constant overpotential of 400 mV in 1 mol/L KOH(B),optimized geometric model of Ni sites in Ni⁃O⁃G SACs structure(C),the corresponding map of the DFT ESP surfaces of Ni⁃O⁃G SACs structure(D),schemeric of oxygen production pathways on the Ni site within Ni⁃O⁃G SACs geometry(E),the free⁃energy diagrams of OER pathways and OER theoretical overpotential of the Ni⁃O⁃G SACs structure(red),Ni⁃N⁃G SACs(pink),and NiO nanoparticles(blue)(F)[69]
3.3 全解水反应
电化学催化全解水涉及到对阳极OER和阴极HER的整体催化.与半反应HER或OER不同,全解水过程要求阴、阳极催化剂在相同的测试条件(温度、酸碱度等)下保持较高的催化活性和稳定性,因此开发双功能催化剂,即在相同条件下同时对HER和OER具有较高的催化活性,是实现电化学全解水的关键[75,76].此外,对基于PEM等固态电解质的膜电极型电解水器件,阳极产生的氧气会在催化材料表面产生活性氧,促进固态电解质降解,加速器件失效[77].降低全解水器件中电极催化材料的ORR活性,可以有效避免活性氧对固态电解质的降解,因此开发低ORR活性、高HER/OER活性的单原子催化材料用于电解水是提高反应效率和器件稳定性的有效手段[78].
通过改变单原子催化材料的配位环境和电子结构来调节吸收能,使得同一催化材料展示出对OER和HER的双功能催化活性[79].Talib等[9]采用第一性原理计算方法,研究了PMA簇支撑的一系列SACs对HER,OER和ORR的电催化性能.结果表明,主要是Pt1/PMA,Ru1/PMA,V1/PMA和Ti1/PMA 表现出较好的HER 催化性能.Co1/PMA(0.45 V)和Pt1/PMA(0.49 V)可以作为高活性和选择性的OER 催化剂.其中Pt1/PMA因其兼具优异的HER和OER活性,有望作为最有发展前景的双功能催化材料被应用于全解水生产中.此外,可以将活性金属通过不同的锚定方式固定在同一催化材料的不同位置处,形成具有不同催化功能的催化材料,最终制备出目标双功能催化材料.Lai 等[10]报道了一种电子辅助策略用来锚定单原子位点,制备了一系列金属(M=Ir,Pt,Ru,Pd,Fe,Ni)单原子催化剂并用于全解水反应.如图6(A)所示,金属单原子分别锚定在N掺杂的碳和CoO八面体中心上,这种独特的结构使得此种单原子催化剂具有同时提高HER和OER催化效率的能力.通过对多种金属单原子催化剂的对比发现,Ir1@Co/NC表现出优异的碱性全解水活性[图6(B)],在10 mA/cm2的电流密度处,其全解水电解电位为1.603 V.计算结果显示,锚定在CoO八面体中心上的Ir单原子结构[CoOIr1@CoO(Ir)]有利于提高OER 性能,而锚定在N 掺杂碳上的Ir 单原子结构(Ir1@NC3)可以加强HER 的催化效率[图6(C)~(E)].Sun 等[80]合成了一种基于Mo 单原子修饰的Co9S8催化剂(Mo-Co9S8@C),该催化剂具有优异的OER 和HER催化活性.在酸性介质中,当电流密度达到10 mA/cm2时,在Mo-Co9S8@C上的OER和HER催化过电位分别为370和98 mV.在碱性介质中,当电流密度达到10 mA/cm2时,Mo-Co9S8@C的OER和HER催化过电位分别低至200 和113 mV.在10 mA/cm2的电流密度处,全解水电解电位在酸性和碱性介质中分别为1.68 和1.56 V.此外,该催化剂表现出优异的催化稳定性.计算结果表明,Co9S8与Mo 单原子之间存在的协同电荷转移耦合效应、高效的电子运输能力以及原子金属与载体之间的强相互作用,提高了该催化材料的稳定性和电催化活性.
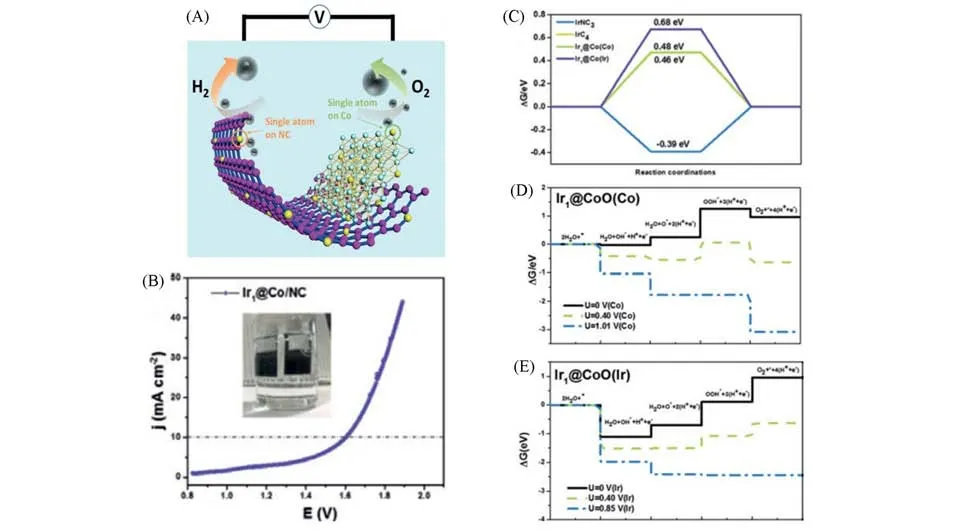
Fig.6 Illustration of the working mechanism of the prepared electrodes(A),the polarization curves of over⁃all water splitting by the Ir1@Co/NC catalyst(B),the free⁃energy diagrams for the HER at pH=14 on IrNC3,IrC4,Ir1@Co(Co),and Ir1@Co(Ir)(C),the reaction free energies for the OER at pH=14 of the intermediates on Ir1@CoO(Co)(D)and Ir1@CoO(Ir)(E)[10]
4 氢燃料电池
氢燃料电池包括阴极ORR 和阳极氢氧化反应(HOR),其中阴极上缓慢的氧还原反应动力学限制了氢燃料电池的广泛应用[11,20,81~85].因此,开发高效稳定的ORR 催化材料是提高氢燃料电池的关键.根据交换膜的不同,可以将氢燃料电池分为阴离子交换膜燃料电池(Anion exchange membrane fuel cell,AEMFC)和PEMFC(图7).由于针对阳极HOR的单原子催化研究相对较少[86],下面主要介绍用于阴极ORR的单原子催化材料在氢燃料电池膜电极器件中的应用.
Fig.7 Schematic diagram of PEMFC(A)and AEMFC(B)
4.1 质子交换膜燃料电池
PEMFC作为一种高效、环保的能源转换技术,有助于缓解环境和能源相关问题[87,88].PEMFC通常采用氢气和氧气作为反应气体,在阳极发生HOR,在阴极发生ORR,其中,ORR因其缓慢的动力学严重影响了电池效率.目前,PEMFC主要采用Pt作为电催化材料,但由于Pt的价格昂贵、资源匮乏,使得催化剂材料成本居高不下,也是限制燃料电池大规模商业化的关键[89,90].因此,开发基于单原子的高性能阴极催化材料已成为重要研究方向[84,88].Xu等[91]报道了一个碳缺陷锚定的Pt SAC,由于其超高的Pt 利用率,在PEMFC 中表现较高的ORR 性能,在H2/O2测试环境中,峰值功率密度为520 mW/cm2.研究结果表明,高效的ORR催化活性主要是由于碳二空位中由4个碳原子锚定的Pt原子结构(Pt-C4).Liu等[92]通过高温热处理的方法制备了单原子Pt催化剂Pt1@Pt/NBP.在酸性介质中,Pt1@Pt/NBP显示出优于商业Pt/C的氧还原催化活性,其半波电位为0.867 V.此外,将Pt1@Pt/NBP作为阴极催化剂应用于质子交换膜燃料测试中,在H2/O2测试条件下,峰值功率密度高达1.14 W/cm2.理论计算发现,Pt1@Pt/NBP优异的氧还原催化活性和全电池性能归因于其独特的活性位点结构,即Pt单原子与一个吡啶氮和两个碳原子配位,该结构可形成四电子ORR路径,进一步提高了ORR性能和全电池效率.Liao等[93]开发了一种g-C3N4驱动的高密度Fe-N4结构的空心碳多面体作为ORR催化剂,并将其应用于PEMFC测试中.在酸性和碱性半电池测试中不仅表现出优异的催化活性,且表现出较高的催化稳定性.H2/O2测试环境中达到628 mW/cm2的峰值功率密度,此外,在8 h 的耐久性测试之后仅有51.3%电流密度损失.研究表明,g-C3N4的存在极大抑制了氧化N的形成,诱导吡啶N富集,促进了Fe-N4结构的形成,从而提高了ORR催化活性.
Xie等[94]通过溶液合成途径将配体螯合的CoNx基团固定在ZIF-8的微孔中,利用ZIF-8独特的碳氢化合物网络,作为单个Co原子之间的保护屏障,降低了它们的迁移率,避免了Co凝聚,从而增加单个Co 位点的密度,最终制得具有非平面类卟啉MN4C12结构高负载的Co 单原子催化剂(Co-N-C)[图8(A)].如图8(B)所示,该催化剂具有较高的ORR 催化活性,其起始电位(Eonset)和半波电位(E1/2)分别为0.93和0.82 V,远高于商业Pt/C催化材料.在H2/O2条件下的PEMFC测试中,峰值功率密度达到0.64 W/cm2[图8(C)].如图8(D)所示,在相同燃料电池测试环境中,对含有Co(mIm)-NC(1.0)和Fe(mIm)-NC(1.0)两种阴极催化材料的燃料电池进行耐久性测试,结果表明,添加Co(mIm)-NC(1.0)的燃料电池在0.7 V下的电流密度下降了20.5%,而使用Fe(mIm)-NC(1.0)的燃料电池的阴极效率为46.8%.对催化降解机制进行研究发现,催化材料的失活主要是由于自由基氧化和活性位点脱金属.如图8(E)所示,CO2气体的生成量和电极电位无关,可以推测大部分CO2由过氧化氢衍生的自由基引起的的化学氧化,而不是电化学氧化,电化学氧化与电极电位有关.在更高的0.85 V电压下进行50 h耐久性测试发现,相比于Fe(mIm)-NC(1.0)阴极催化剂,Co(mIm)-NC(1.0)阴极催化剂的膜电极组件(MEA)更耐用[图8(F)].测试了Co(mIm)-NC(1.0)和Fe(mIm)-NC(1.0)在测试过程中的脱金属量[图8(G)],无论在何种条件下,被浸出的Co的含量均低于被浸出的Fe的含量,特别是在80 ℃,O2净化的0.5 mol/L H2SO4溶液中循环之后,被浸出的Co 要比Fe 少得多(7.3%Covs.40%Fe).此外,DFT结果表明,该催化剂中较短的Co—N键长(0.194 nm)可以降低Co金属的溶出[图8(H)和(I)].
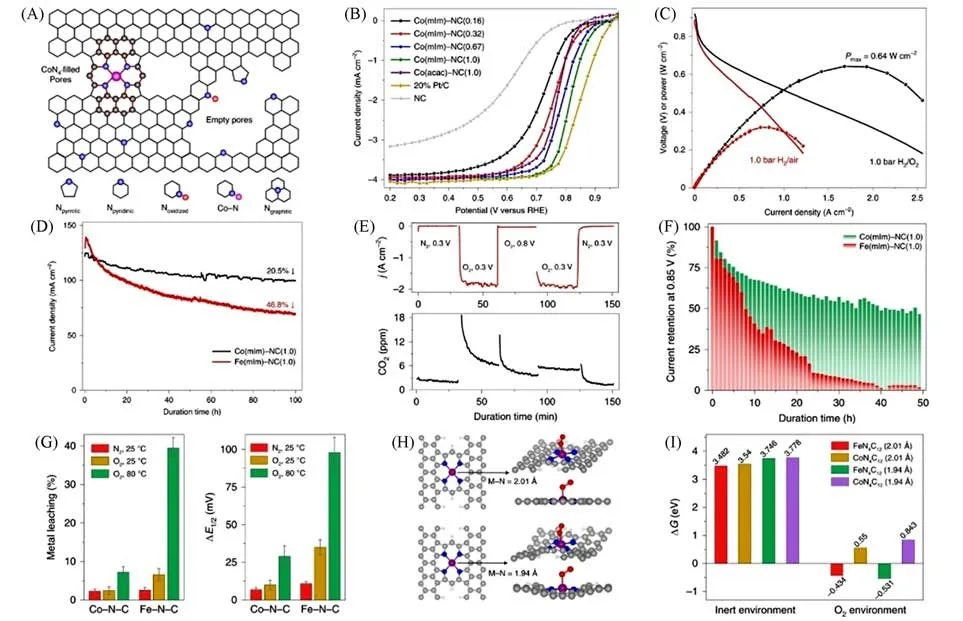
Fig.8 Plan view of presumed porphyrin⁃like CoN4C10 sites in the micropores(A),ORR polarization plots for different Co(mIm)⁃NC catalysts in 0.1 mol/L HClO4 under O2 saturation(B),fuel cell performance of Co(mIm)⁃NC(1.0) measured under 1.0×105 Pa H2/O2 and H2/air(C),durability tests of the Co(mIm)⁃NC(1.0)and Fe(mIm)⁃NC(1.0)catalysts in MEA in 1×105 Pa H2/air at a con⁃stant cell voltage of 0.7 V for 100 h(D),in situ CO2 emission test from the fuel cell cathode with the Co(mIm)⁃NC(1.0)catalyst(E),normalized current density at a voltage of 0.85 V after voltage⁃step cycling(0.4 V for 55 min and 0.85 V for 5 min) for 50 h(cycles) measured under 1.0×105 Pa H2/O2(F),ICP⁃OES and RRDE(E1/2) results of the metal leaching experiments(G),atomistic struc⁃tures of simulation models of porphyrin⁃like MN4C12(M=Fe or Co) active sites and their corre⁃sponding O2 adsorption configurations on the M site(H),predicted thermochemical constants of losing an M atom from the porphyrin⁃like MN4C12(M=Fe or Co)sites(I)[94]
4.2 阴离子交换膜燃料电池
与商业化PEMFC 相比,AEMFC 的电化学催化过程是在碱性环境中进行的,从而避免了对金属活性位点的强腐蚀[95~97].此外,AEMFC具有较高的峰值功率密度(Maximum power densities,Pmax),目前报道的AEMFC 峰值功率密度已高于2 W/cm2[98,99].已报道的具有高效率的AEMFC 需要在阴阳两极负载高载量的Pt催化材料,为AEMFC进一步商用化带来极大的障碍.从长远看,开发用于AEMFC的非Pt催化材料成为解决提高电池效率的关键问题之一[100].对于阴极而言,碱性环境下的ORR 比酸性环境下更容易发生,因此可以使用非铂族金属(non-PGM)作为ORR活性中心.这为非贵金属单原子催化材料作为ORR催化材料在AEMFC中的应用提供了可能[96,101].Zhou等[102]通过配位辅助聚合组装方法,制备了一种独特的Fe 分散N 掺杂介孔碳球(meso-Fe-N-C).meso-Fe-N-C 在0.1 mol/L KOH 溶液中的ORR半波电位为0.846 V,动力学电流密度(Jk)为4.696 mA/cm2,催化活性远远优于商业Pt/C.将制备的单原子催化材料制成膜电极应用于阴离子交换膜燃料电池测试,发现该催化剂具有优异的全电池催化活性,在H2/O2测试条件下,其峰值功率密度达到584 mW/cm2.Xiang 等[103]通过热解的方法制备了Cu SAC,并将其作为AEMFC 阴极催化材料,在H2/O2测试环境中,功率密度达到196 mW/cm2.研究表明,Cu 单原子位点有利于降低超氧羟基自由基(OOH*)向氧自由基(O*)转化时的吉布斯自由能(ΔG).Liu 等[101]制备了一种具有Fe-N5活性位点的N,F 双掺杂的多孔碳纳米材料C@PVI-(DFTPP)Fe-800,该材料表现出优异的碱性ORR 催化活性,其半波电位达0.88 V,转化效率为0.47 e·site-1·s-1.作为AEMFC的阴极催化材料,该催化材料表现出优异的全电池性能,其开路电压为0.93 V,峰值功率密度为104 mW/cm2.
Adabi等[97]通过增加材料的平均孔径和石墨化水平改良了传统Fe-N-C催化剂,获得了一种高效的氧还原催化材料,并将其作为ORR 催化剂应用于AEMFCs 的阴极催化反应中.如图9(A)所示,在低温度和全湿度(阳极/阴极/电池温度为60/60/60 ℃)下工作时,电池性能较低,因为阳极处有大量的积水.在电池温度为70 ℃时,当阳极/阴极露点从60/64 ℃降到60/62 ℃时,峰值功率密度从1.4 W/cm2增加到1.6 W/cm2.在先前80 ℃的最佳露点下,施加200 kPa 的背压.结果是极高的峰值功率密度为2.05 W/cm2,在0.1 V 时电流密度为7.4 A/cm2.如图9(B)所示,在600 mA/cm2下进行耐久性测试,发现该AEMFCs表现出优异的耐久性,在150 h后基本保证各参数不变.对比分析了已报道的不同催化材料,表明该Fe-N-C催化材料表现出优于其它催化材料的AEMFCs效率[图9(C)].
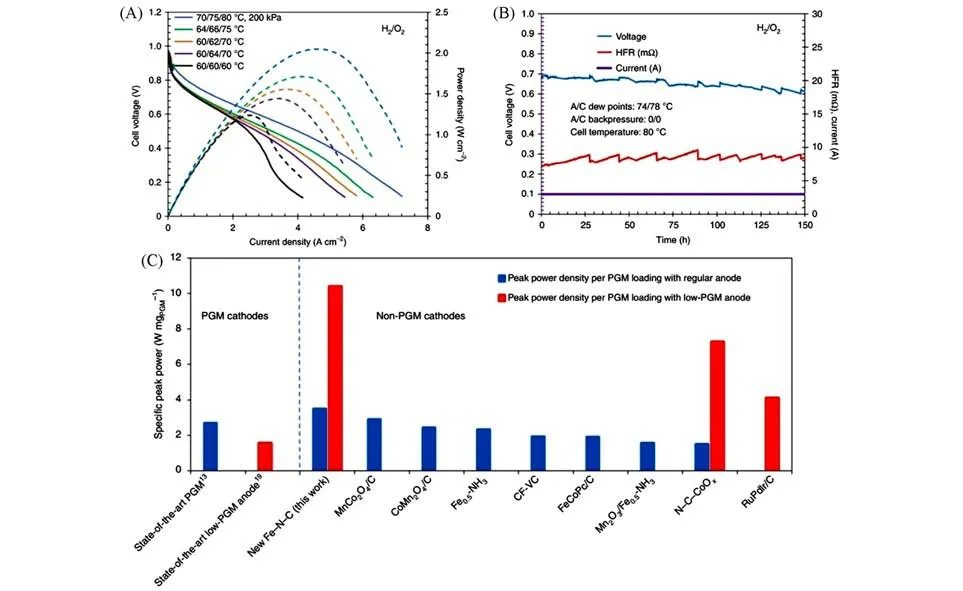
Fig.9 Current density⁃voltage(solid) and current density⁃power density(dashed) curves for a H2/O2 AEMFC with a cathode with 1 mg/cm2 Fe⁃N⁃C,an anode with 0.6 mg/cm2 PtRu(A),voltage,HFR and current versus time curves recorded at a constant current density of 600 mA/cm2(B),compari⁃son of the specific peak power(W/mgPGM loading)with H2/O2 for different AEMFCs,both state⁃of⁃the⁃art PGM⁃type cells and those employing PGM⁃free electrocatalysts(C)[97]
基于非贵金属的单原子催化材料应用于燃料电池,相较于Pt/C基催化材料有显著的成本优势,近年来在性能方面也逐步展示出潜力[104].随着研究的进一步深入,逐步发现将单原子催化材料作为阴极催化层(Cathode catalyst layer,CCL)应用于PEMFC 仍面临诸多科学和技术方面的挑战.其中,因其单位点活性较低,且金属位点密度难以显著提升,使得基于单原子催化材料的CCL层较厚,将严重限制催化过程中的传质效率[105,106].此外,单原子催化材料表现出较差的耐久性,主要是由催化材料中脱金属化、自由基攻击、活性部位质子化作用/阴离子绑定和微孔堵塞等原因引起[104].因此,可从提高金属单原子的载量和优化CCL结构等方面,开发基于单原子催化材料的高效稳定催化层技术.
5 总结与展望
从单原子催化材料在电催化氢气循环反应中的应用出发,系统总结了单原子的结构设计和功能设计策略,以及在电解水制氢和氢燃料电池领域的应用.此外,总结了常用的电化学评价参数.
理论上,单原子催化材料具有接近100%的原子利用率,对降低贵金属成本,提高催化效率具有十分重要的意义.同时,在结构上单原子催化材料作为一类仅含有孤立的单个金属原子作为催化活性中心的负载型催化材料,打通了多相催化到均相催化的桥梁.此外,可以通过对金属单原子周围的化学环境进行调控,实现具体催化反应的需求.对单原子催化活性中心的微观调控,主要是基于调节活性中心的空间结构以及电子分布,常用的方法包括:(1)配位效应:通过改变金属单原子周围的杂原子(B,C,N,P,S等)改变催化位点的配位结构,实现催化活性提升;(2)空位效应:该方法是将具有原子空位的材料(如C空位、含O空位的金属氧化物等)作为载体,将金属单原子锚定在原子空位处,达到稳定金属单原子的目的,同时金属和载体间的协同作用也在一定程度上对催化反应有提升作用;(3)掺杂策略:主要是将特定金属单原子掺入到某些金属氧化物、磷化物、硫化物等晶体结构中,被掺入的金属单原子取代原有金属原子存在于晶格中,该方法制备的单原子催化材料一般不会破坏原有的晶体结构,在提升催化活性的同时,保证了结构的稳定性;(4)离子交换策略:一般是指MOF基单原子催化材料,由于ZIF-8中的Zn原子在高温条件下被蒸发,目标金属原子取代Zn原子的位置,形成单原子催化材料.
将合成的单原子催化材料应用到实际的催化反应和电化学器件中,并最终达到商业化是单原子催化研究的终极目标.本综述对单原子催化材料在电解水制氢及氢燃料电池领域的应用进行了总结.
在电解水制氢中,阳极OER反应较高的催化过电位以及反应活化能制约了全解水的进一步应用.特别是,高电位下催化材料的活性和稳定性问题.为了解决这一问题,可以通过以下手段来进行催化材料的调控:(1)通过单原子金属与载体之间的复合,实现单原子与载体之间的协同,达到提升催化活性的同时,提高了催化材料的结构稳定性,进一步提升催化稳定性;(2)通过调配单原子周围的配位原子,实现金属与周围环境的电子耦合,改善催化活性位点对中间产物的吸脱附能,提高催化活性,降低中间产物对活性位点的毒化,同时保证催化活性和耐久性;(3)设计双金属或多金属单原子催化材料,利用不同金属的化学属性构建级联催化材料,针对性地对多电子反应中的各个中间产物进行分级催化,达到提高催化活性、保证催化稳定性的目的.
在燃料电池中,商业Pt/C作为阴极ORR催化材料仍具有无可取代的地位,而非贵金属单原子催化材料(如Fe-N-C等)在质子交换膜燃料电池中容易发生金属溶解,使其催化活性迅速下降,限制了非贵金属单原子催化材料在PEMFCs中的实际应用.与PEMFCs相比,AEMFCs 超高的pH环境避免了对金属活性位点的腐蚀,使得非贵金属催化材料在AEMFCs的实际应用成为可能.此外,除了急需解决的催化材料的问题,阴离子交换膜的制备也是AEMFCs开发研究中的核心问题之一.
综上,通过单原子催化材料的结构和功能设计,满足对不同催化反应的高效稳定催化需求,推动单原子催化材料在电化学全解水和氢燃料电池中的实际应用,以实现单原子催化在电化学氢循环领域的发展.单原子催化材料在实际应用中的核心瓶颈是稳定性问题,为了进一步解决单原子催化材料的耐久性问题,或可尝试从以下两个方向开展研究:(1)通过金属掺杂制备合金化的单原子催化材料,合金化的过程即保证催化活性的提升,同时增加了催化材料的稳定性.合金化的单原子催化材料有利于贵金属的合理有效利用,大大提高了原子利用效率.此外,单原子活性中心独特的电子结构和界面处不同金属间的相互作用达到协同催化的效果,从而提高催化材料的活性和稳定性[107].(2)采用分子封装的手段,降低金属活性位点的腐蚀,避免催化活性位点的坍塌,在保证催化活性的同时,增强催化稳定性.如,Chen等[108]采用环糊精作为封装小分子,对单原子Fe活性位点进行局部封装,不仅有效降低了Fe单原子的溶解,还有效阻挡了OH-对Fe活性位点的过量吸附,既提高了Fe单原子催化剂的活性也保证了催化剂的耐久性.此外,针对单原子催化的研究集中在三电极体系的半电池反应,而电解水和燃料电池的基元器件中存在荷质传输、水-热管理和微多相流等科学问题,存在多样化的失效机制,综合性能(催化活性、材料稳定性)与半电池评价结果存在一定差异.因此,在基本物化性能和催化性能达到应用要求的前提下,应进一步在器件中评价其综合性能,解决应用中的基础科学问题.
