自支撑单原子膜电极在能源电催化中的应用
2022-06-29何观朝费慧龙
吴 俊,何观朝,费慧龙
(湖南大学化学化工学院,化学生物传感与计量学国家重点实验室,长沙 410082)
传统化石能源的枯竭问题及伴随的环境污染问题使得改进传统能源的利用方式及寻找新能源作为替代品变得迫在眉睫,而电化学反应与电催化剂在这一过程中将扮演重要角色.单原子催化剂由于100%的理论原子利用率以及基于其独特电子结构所展现出的高催化性能[1],在能源电催化领域引起广泛关注.本文将自支撑单原子膜电极定义为自支撑导电膜片基底负载的金属单原子材料,其显著特征为:宏观上,材料形状为膜片状,而不是粉末状(颗粒状);在电催化应用中,可直接作为工作电极,不添加黏结剂和导电助剂.
由传统粉末状电催化剂所制备的浆料涂层电极存在一些固有的缺点.(1)粉状材料一般依赖于绝缘黏结剂涂覆于集流体而形成电极,导电性较差,部分活性位点被覆盖,催化性能下降.并且有些材料本身导电性差,因而需要加入导电助剂以增强电极的导电能力.但黏结剂和导电助剂的引入增加了“死体积”,不可避免地提高了非活性部分的占比,影响电极材料的催化性能[2~5].(2)粉末状电催化剂材料与衬底之间黏合力较弱,同时为保障电荷传导顺畅并防止材料层出现断裂、分层的问题,电极厚度不能过大,催化剂的载量通常小于1 mg/cm2,进而导致活性位点数量有限[6~8].(3)在长时间或大电流工况条件下,电解液中反应物与产物的传质受限问题愈发明显,气体产物的脱离更加困难,导致黏附于集流体上的催化剂材料容易发生脱落[9].因此,大多数的粉末状电催化剂仅在电流较小(<100 mA/cm2)、工作时间较短条件下进行性能评估[10,11],无法达到工业应用的要求.
与传统的粉末状电催化剂相比,自支撑单原子催化材料具有独特的优势.首先,整体式的电极结构更加稳固,各部分之间“无缝衔接”构成一体化的导电框架,有利于电荷的传输[5].其次,自支撑基底锚定、分散金属单原子,可防止其团聚成为纳米颗粒.基于活性单元原位生长的特点,活性位点被覆盖、包埋的概率也小于粉末催化剂所形成的涂层电极,因而可暴露更多活性位点,展现出优异的催化性能.对比之下,粉末材料(尤其是高负载量下)活性位点的利用率较低[12~14].最后,通过调控自支撑材料的形貌与孔结构,可以实现一些特殊的功能,例如特殊的孔道结构可提升传质能力[10]、材料表面疏气性的调控可加速气体产物脱离电极等[15].基于以上优点,自支撑单原子材料在大电流电化学反应、高能量高功率密度电池中具有良好的应用前景.
迄今,关于自支撑催化剂的研究已有一些报道[5,9,16~18],但多侧重于以纳米颗粒为催化单元的自支撑材料,对于以单原子为催化活性位点的自支撑电极的综述报道较少.本文聚焦于自支撑单原子催化剂的一些合成方法与策略,基于其单原子活性位点及自支撑材料结构的特点,对其在能源电催化中的应用进行了评述.
1 自支撑单原子催化膜电极的合成方法
1.1 自支撑基底上原位制备单原子材料
以碳布(CC)、泡沫镍(NF)、泡沫铜、纳米纤维及金属化合物等为基底制备自支撑结构材料已有较多报道[18].通过合适的方式在上述基底中引入单原子位点是合成自支撑单原子催化剂的一种常用手段.
Xie 等[12]以CC 为基底制备了自支撑钴单原子催化剂[SS-Co-SAC,图1(A)].他们在含有硝酸锌、硝酸钴和2-甲基咪唑的混合溶液中放入经高锰酸钾溶液刻蚀的碳布,静置2 h,使锌钴-沸石咪唑骨架(ZnCo-ZIF)纳米片阵列在碳布表面密集生长.由于锌、钴元素与2-甲基咪唑具有相同的配位结构,锌离子的过量引入导致钴离子以孤立状态存在.高温热解过程中,孤立的钴离子通过周围有机配体的还原及锌原子蒸发产生的缺陷锚定,原位形成钴单原子.高温热解前后,CC表面的纳米片阵列结构保持完好,仅有轻微收缩.透射电子显微镜(TEM)图像显示超薄纳米片上存在大量的介孔,原子力显微镜图像显示纳米片的厚度约为35 nm,原子级分辨率的高角度环形暗场扫描透射电子显微镜(HAADFSTEM)照片证实了钴金属以单原子形式高密度地分散在纳米片上.
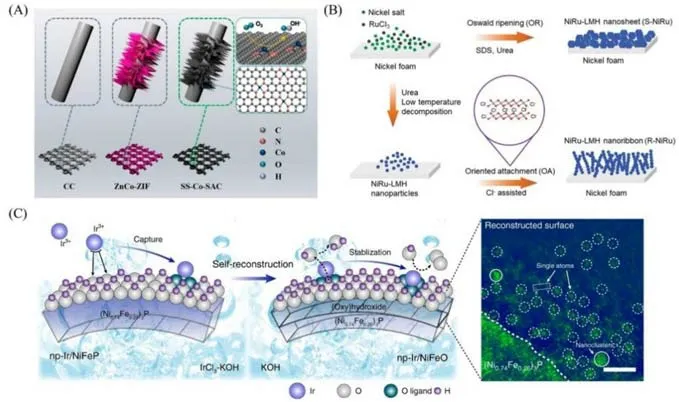
Fig.1 Schematic illustrations for the synthesis of SS⁃CoSAC(A)[12],NiRu⁃LMH ultrathin nanoribbons(R⁃NiRu)and nanosheets(S⁃NiRu)(B)[20],and np⁃Ir/NiFeO(C)[23]
层状金属氢氧化物(LMHs)中大量带负电荷的羟基官能团对其表面的纳米材料具有限域作用,同时表面的羟基可锚定单原子,是制备单原子催化剂的良好基底材料[19].但作为一种典型的二维材料,LMHs 趋向于片状生长,大部分活性位点存在于片层之间,原子载量的提升受到限制.鉴于此,Chen等[20]以NF为基底制备了负载在超薄氢氧化镍纳米带上的自支撑钌单原子催化剂[图1(B)].他们将经预处理的NF浸于含有NiCl2·6H2O,CO(NH2)2,RuCl3·H2O和NaCl的混合溶液中,通过水热处理合成了目标材料.在此过程中,氯离子作为结构导向剂,对于超薄纳米带结构的形成起到关键作用;与之对应,在溶液中仅有少量氯离子、更高水热温度的条件下,未观察到纳米带结构的形成.基于纳米带的结构优势,实现了钌原子的高负载量(质量分数约为7.7%).以NF为生长基底,保证了自支撑材料的导电性与稳定性;纳米带相互交织形成网络结构,在保证机械性能的同时,层级的间隙和孔隙结构可提供大量的传质路径.以泡沫铜作为基底制备自支撑单原子材料也有报道,例如,Liu等[21]通过溶剂热法在泡沫铜上生长了WO3阵列,用于负载钴单原子催化剂.
过渡金属化合物,如金属氧化物(或金属氢氧化物)、金属硫化物、金属磷化物、金属氮化物、金属硒化物等,对于特定的电化学反应具有一定的催化活性[22].将其作为基底负载金属单原子,催化性能可得到提升;对于贵金属,以单原子位点形式存在可极大减少其使用量.Jiang等[23]使用一种简单的自重构策略在自支撑多孔(Ni0.74Fe0.26)3P基底(np-NiFeP)上可控沉积铱单原子[图1(C)].自支撑np-NiFeP基底通过电化学选择性刻蚀制备[24],采用循环伏安法将铱离子原子级沉积至基底,合成前驱体材料np-Ir/NiFeP.在碱性溶液中经电化学活化后,前驱体表面发生自重构,生成无定形的镍铁氢氧化物层,铱单原子被氧化,价态更高,与更多氧配体配位,处于更加稳定的状态,最终形成催化剂np-Ir/NiFeO,适应更加极端的反应条件.Jiang等[22]通过电化学沉积的方式,在自支撑多孔Co0.85Se基底上合成了铂单原子催化剂.
在自支撑基底上合成单原子催化剂在一定程度上保障了所制备材料的机械性能和导电性.但自支撑基底的合成较复杂,需要寻求简易、低成本的自支撑基底材料制备方式并通过选择合适的方法引入单原子位点,以确保单原子活性位点稳固地与基底结合,保障其电化学稳定性.
1.2 静电纺丝法
静电纺丝是利用静电力从聚合物溶液或熔体中合成纳米纤维的独特技术,其设备主要由高压电源、喷丝头和收集器三部分组成.在高压电场作用下,聚合物溶液或熔体以带电射流形式从针头喷出,从“泰勒锥”伸长,依赖于所带电荷的排斥力形成长细线,待溶剂挥发后得到固化纤维[25].通过调节喷丝头与收集器可有效调控纤维的结构,实现纳米纤维的扭曲结构、核壳结构、中空管状结构、多孔结构和分级结构[25,26],且纳米纤维可随机排列或定向排列形成纤维膜[27].静电纺丝制备的纳米纤维碳膜具有化学成分可控、孔隙率高、比表面积大、机械强度好及导电性能优良等优点[28],是理想的自支撑电极材料.
Yang 等[29]以吸附铜离子的ZIF-8和聚丙烯腈(PAN)为前驱体,通过静电纺丝法及相应后处理,合成了负载铜单原子的碳纳米纤维膜(CuSAs/TCNFs).组成碳膜的碳纳米纤维相互交联形成直径为700 nm左右的网络结构,且其本身具有孔径均一(约100 nm)的贯通纳米孔结构.值得注意的是,在保持铜离子含量不变的前提下,将ZIF-8量降至原来的三分之一,碳纳米纤维的纳米孔结构显著减少且相互之间不连通;而在不加入铜离子,仅以ZIF-8 和PAN 为前驱体条件下,碳纳米纤维却保持了与CuSAs/TCNFs类似的贯通纳米孔结构,证明纳米孔结构的形成与ZIF-8密切相关.Yang等[30]使用静电纺丝技术制备了高产量、强韧性的自支撑镍单原子多孔碳膜材料[NiSA/PCFM,图2(A)].由ZIF-8、镍盐[Ni(NO3)2·6H2O]和PAN组成聚合物溶液,ZIF-8承担造孔功能,镍单原子由镍盐引入.通过静电纺丝技术,将聚合物溶液合成为纳米纤维;接续的高温热解使得镍金属以原子级形式分散,锚定到氮掺杂多孔碳基底上.最终,自支撑镍单原子多孔碳膜材料的尺寸至少为280 cm2,适宜较大尺寸或厚度薄膜的制备[图2(B)].薄膜机械性能良好,具有柔性,在弯曲应力作用下仍能保持初始结构,能承受约1.3 MPa的拉伸强度[图2(C)].
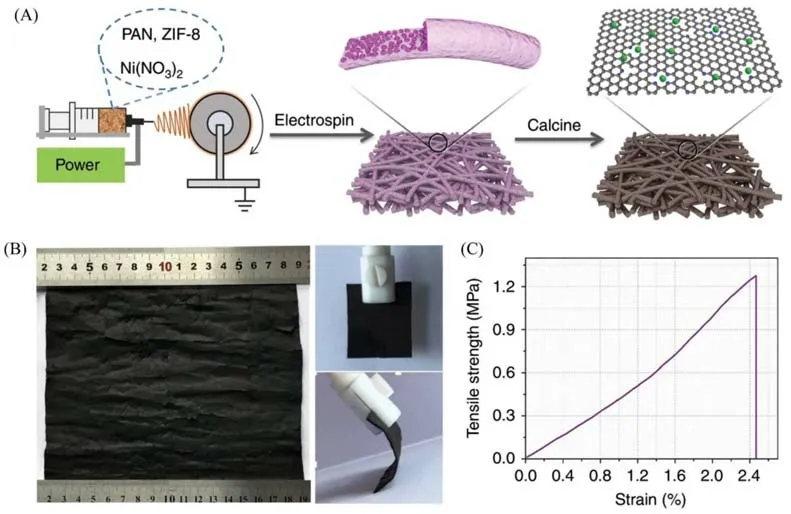
Fig.2 Synthetic strategy(A),digital images(B)and stress⁃strain curve(C)of NiSA/PCFM membrane[30]
1.3 自组装法
自组装是一种基本结构单元自发形成有序结构的技术.在自组装过程中,基本结构单元在非共价键相互作用下自发地组织或聚集为一个稳定、具有一定规则几何外观的结构.通过自组装制备自支撑单原子材料的优势在于合成工艺相对简单,能耗较小.
Liu 等[10]报道了一种定向多孔氮掺杂碳膜负载的钴单原子材料[Co-NG-AF,3(A)].含有乙醇、钴盐及高氧化程度多壁碳纳米管(O-MWCNTs)的悬浮液首先经过水热过程自组装形成水凝胶,经过接续的液氮定向、冷冻干燥处理,水凝胶变为定向多孔的气凝胶[图3(B)].乙醇对于定向效果具有显著作用,其加入降低了冰点,影响冰晶生长的结晶行为.冷冻诱导的相分离促使O-MWCNTs沿着冰晶生长方向堆积,冷冻干燥去除冰模板后得到定向多孔气凝胶.最终,在Ar/NH3气氛中退火处理,合成了Co-NG-AF催化材料.在碳膜表面,孔径为10~20 μm的孔均匀分布[图3(C)],截面是长程有序的竖直定向排列孔道,孔道直径约为20 μm[图3(D)].

Fig.3 Synthetic route(A),optical image(B),top⁃view SEM image(C) and cross⁃sectional SEM image(D)of Co⁃NC⁃AF[10]
Xing等[31]以胺功能化氧化石墨烯和钴盐为前驱体,通过水热自组装、定向冷冻及超快焦耳热处理等步骤制备了具有三维多孔整体结构的单原子钴氮掺杂石墨烯材料.基于焦耳热处理瞬态淬火的特点,材料避免了额外加热诱导的原子聚集,使得原子尺度的CoNx活性位点快速、稳定地分散于石墨烯基底.石墨烯片层在自组装过程中形成了相互连接的大孔和纳米孔,在催化过程中,有利于离子和气体的畅通传输.
Li 等[32]使用层层组装的方法制备了自支撑钴单原子材料.他们在纤维素纤维(CNF)和碳纳米管(CNT)混合溶液中加入负载钴单原子的氮掺杂碳纳米片(Co@NC),混合均匀后,通过真空泵抽滤,获得自支撑的CNT-CNF-Co@NC膜.其厚度约为10 μm,具有优异的机械性能,TEM图像显示,Co@NC紧密地锚定于CNT-CNF复合网络中.
1.4 化学气相沉积与固相扩散法
非均相催化剂等固体材料的微观结构决定了与尺寸、形状相关联的性能.在纳米甚至原子水平上调控固体材料的结构会使局部化学环境发生极大改变,从而提升相应的性能[33,34].固相扩散常用于调控材料的微观结构,以获得特殊的化学和物理性质.Zhao等[15]直接以块状金属为原料,通过固相扩散合成自支撑单原子材料[图4(A)].他们利用喷涂设备将三聚氰胺沉积至镍箔表面,形成三聚氰胺薄膜.通过精准地控制加热速率,三聚氰胺逐渐转化为C3N4结构,覆盖于镍箔表面.在高温下,经镍源的催化,C3N4结构转化为氮掺杂的碳膜(N-C).在Ni-N配位的强路易斯酸-碱作用驱动下,形成大量碳空位,镍箔表面的镍原子扩散进入N-C膜中,同时碳流扩散至镍箔,形成反向流动相平衡.在N-C膜中,镍纳米颗粒作为“种子”,氮掺杂碳纳米管(N-CNTs)以“气-液-固”机制生长,当镍纳米颗粒周围碳过饱和时,“碳流”析出,N-CNTs生长.C3N4在持续高温下蒸发,不断为N-CNTs的生长提供碳源和氮源,在两侧生长出垂直于二维N-C膜的一维N-CNTs,形成分级结构.最后,将其从镍箔上剥离,经过酸浸法溶解大部分镍金属纳米颗粒,得到具有分级结构的自支撑镍单原子碳纸材料(H-CPs).使用不同形状的镍箔,可制备相应形状的碳纸材料,其柔韧性、稳固性与商用碳布一致,抗拉伸强度在6 MPa以上.
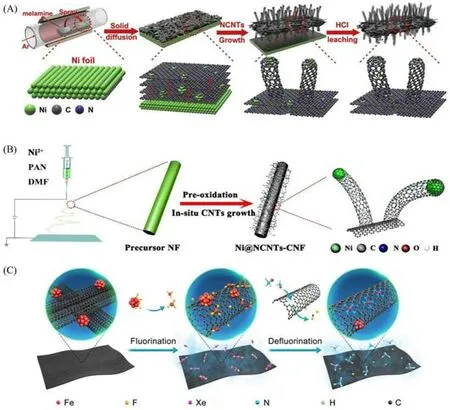
Fig.4 Scheme for the synthesis of H⁃CPs(A)[15],Ni@NCNTs⁃CNF catalysts(B)[35]and the SAFe⁃SWCNT film(C)[36]
Shen 等[35]采用类似的固相扩散方式,在静电纺丝制备的碳纳米纤维膜上原位生长了N-CNTs[图4(B)].镍单原子、镍纳米颗粒分别是优异的二氧化碳还原反应(CO2RR)和析氢反应(HER)活性位点,合成的催化剂(Ni@NCNTs-CNF)含有双活性位点.不同于以块状金属作为金属源,该研究以Ni(acac)2作为金属源,依靠碳纳米纤维膜的预氧化处理,促进镍前驱体向纤维表面扩散,将“种子”布于纤维表面.在后续热解过程中,镍的氧化物还原为纳米颗粒,成为“种子”,N-CNTs原位生长.但固相扩散在高温剧烈反应条件下对于微观结构的控制能力较差,不是一种有效、高效的复杂原子结构固体材料制备方法.
Meng等[36]分别以二茂铁和甲苯为催化前驱体和碳源,噻吩为生长促进剂,制备了单壁碳纳米管薄膜(SWCNTs)负载的铁单原子催化剂[图4(C)].首先,通过浮动催化剂化学气相沉积法合成SWCNTs[37],铁金属以纳米颗粒形式附着;然后,进行氟化处理,由于氟的强电负性,将单个铁原子从纳米颗粒中分离,铁原子被氟原子固定并分散在F-SWCNTs上;最后,在氨气环境中高温热解,使材料脱氟,形成空穴,铁原子与氮原子形成配位结构,填入空穴,形成铁单原子催化剂.
Du 等[38]通过化学气相沉积法,以负载了SiO2纳米颗粒的NiMoO4纳米纤维为模板,制备了自支撑三维多孔氮掺杂石墨烯负载的钼单原子/团簇材料.在化学气相沉积过程中,NiMoO4纳米纤维还原为多孔NiMo合金,氮掺杂石墨烯在其表面生长,SiO2纳米颗粒的存在使得形成的纳米管石墨烯产生纳米孔,通过简单地改变SiO2纳米颗粒的使用量可调控石墨烯上纳米孔的数量.化学气相沉积过程后,对合成的一整片材料进行选择性刻蚀,去除模板及SiO2纳米颗粒.该研究中,通过SiO2纳米颗粒的造孔作用在石墨烯基底上引入边缘/缺陷,发现吡啶氮和钼单原子的掺杂水平显著提高.因此,通过造孔等方式增加基底材料的边缘/缺陷位点是提升金属单原子负载量的一条途径.
1.5 其它方法
木材具有天然的分级结构,其连通且有序的孔道结构是理想的传质通道[39,40].Zhong等[41]发展了一种以木材为前驱体、具备规模化制备潜力的自支撑单原子催化剂合成策略.材料的合成主要分为两步[图5(A)]:路易斯酸浸泡处理和高温热解处理.首先,将预处理后的天然木材晒干,置于氯化铁溶液中浸泡.氯化铁会水解木材的部分纤维素和半纤维素,形成大量的纳米孔,同时在木材的孔道中引入了大量的铁离子.然后,在Ar/NH3混合气氛中高温热解,原位形成铁单原子位点.再经盐酸洗涤刻蚀去除铁纳米颗粒后,得到了自支撑单原子材料SAC-FeN-WPC.扫描电子显微镜(SEM)、X 射线能谱(EDS)和球差校正HAADF-STEM表征结果表明,材料中存在孔径小于10 μm的贯通孔道,铁元素均匀分布于基底,且以单原子的形式存在[图5(B)~(D)].
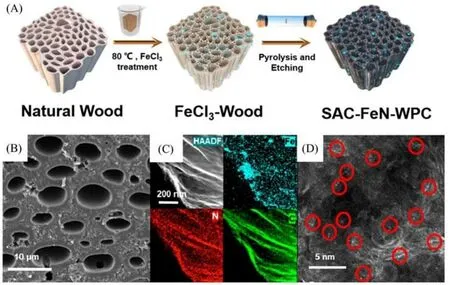
Fig.5 Schematic illustration of the fabrication procedure(A),SEM image(B),HAADF⁃STEM images and the corresponding EDS mapping images(C),Aberration⁃corrected HAADF⁃STEM images with the bright points representing Fe atoms(D)of the SAC⁃FeN⁃WPC[41]
上述的自支撑单原子膜电极合成方法均有独特的优势,也有各自的缺点.在自支撑基底上原位制备单原子催化剂,能最大程度利用已有的自支撑基底材料,并且在一定程度上保证了自支撑单原子膜电极的机械性能及导电性能;但基底结构在后续处理中几乎不会发生明显变化,基底结构的可调控性差,稳定单原子位点的引入有一定难度.通过静电纺丝法制备自支撑单原子膜电极,基底结构、形貌可实现精确调控,膜电极柔性好,可弯曲,具备大规模制备潜力;但聚合物溶液中用于形成纤维的聚合物占比较小,纺丝效率低,有机溶剂在纳米纤维形成过程中挥发,且很难回收,在污染环境的同时也增加了成本.设备要求低、工艺简单、能耗小是自组装法的突出优势,其不足在于不能直接干预基底的形成,无法制备复杂结构的基底.化学气相沉积法可制备各种组分的薄膜,借助模板可以合成复杂结构的基底;但其反应过程温度一般较高,沉积速度慢,最终的膜电极不耐弯曲.固相扩散法常用于材料微观结构的调控,但在较高温度和剧烈反应条件下对结构的控制能力差.
2 能源电催化应用
2.1 析氢反应
氢气拥有142 MJ/kg的高质量能量密度,是汽油的3倍以上,是一种理想的可再生清洁能源[42].目前大部分氢气都是通过化石燃料的蒸汽重整来生产,该种制备方式消耗化石燃料、转化率低,同时又产生二氧化碳.电解水产氢是清洁、可持续的制氢方式,反应物与产物均无污染,但需要高效、稳定的催化剂参与.
Zhang等[43]通过电化学沉积的方法,在生长了磷化钴纳米管阵列(NT)的NF上引入铂单原子,制备了自支撑的铂单原子催化剂(PtSA-NT-NF),并在中性溶液中对其催化析氧反应(HER)性能进行测试.结果表明,当电流密度(jHER)为10 mA/cm2时,其过电位(η)为24 mV,仅比商用铂碳材料(Pt/C)大7 mV,且当jHER>43 mA/cm2时,其η值显著小于Pt/C的[图6(A)],即在较高的电流密度下,PtSA-NT-NF拥有更好的催化性能.当η=50 mV 时,除去NT 和NF 的贡献,Pt 质量活性为70 A/g,是Pt/C 的4 倍;Tafel斜率为30 mV/dec,与Pt/C一致;稳定性优于Pt/C.性能的提升主要归因于单分散高暴露率的Pt原子提高了金属利用率、基底材料对铂单原子电子结构的调控以及无聚合物黏结剂的影响.研究侧重于讨论Pt 金属存在形式的差异(单原子、纳米颗粒)对HER 性能带来的影响,而CoP 的纳米管阵列结构[图6(B)]对HER 性能应该同样有提升作用.在较高的电流密度下,PtSA-NT-NF 拥有更好的催化性能;在更高电流密度下(jHER>80 mA/cm2),NT-NF的过电位甚至比Pt/C更小[图6(A)].显然,阵列结构可有效提高HER性能.NT-NF电化学活性面积大于Pt/C,意味着暴露更多的活性位点.
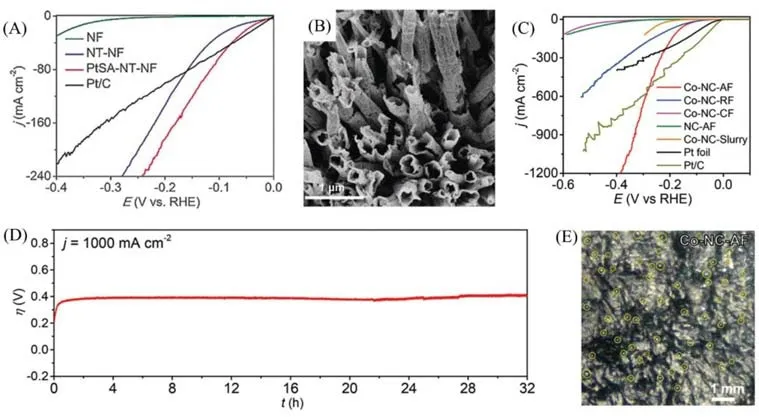
Fig.6 HER polarization curves of different catalysts,acquired with 5 mV/s in N2⁃saturated 1.0 mol/L PBS(A),SEM images of PtSA⁃NT⁃NF(B)[43],linear sweep voltammetry(LSV)curves in 0.5 mol/L H2SO4 at the scan rate of 5 mV/s for different catalysts along with Pt foil and Pt/C as reference point(C),stability of Co⁃NC⁃AF evaluated by the galvanostatic technique(D)and snapshot of H2 bubbles detaching from Co⁃NC⁃AF(E)at the current density of 100 mA/cm2[10]
Liu等[10]制备了一种定向多孔氮掺杂碳膜负载的钴单原子催化剂(Co-NC-AF),更加关注对基底材料结构的设计与表面改性.薄膜电极在500和1000 mA/cm2的电流密度下,过电位分别为272和343 mV[图6(C)],并且能在1000 mA/cm2电流密度下连续稳定运行32 h 以上[图6(D)],实现氢气的快速制备.薄膜电极优异的性能源于材料结构的独特设计及表面改性:具有大比表面积和多级孔隙结构的厚电极可暴露大量的活性位点,提供丰富的“固-液-气”三相界面;贯通薄膜竖直方向排列的微通道及碳壁上的纳米孔保证了电解液与气体产物的通畅传输[图3(D)];材料表面的超亲水性有助于电解液的浸润;亲水性与多孔结构带来的超疏气性使得氢气气泡在体积较小时就脱离材料表面[图6(E)],不会遮挡固-液界面,减少给电极局部带来的应力.与之形成鲜明对比的是,如图6(C)所示,在大电流密度下,由于传质受限且气泡脱离材料带来局部应力可能造成的材料脱落[44,45],使得氮掺杂碳负载的钴单原子粉末催化剂(Co-NG-Slurry)仅能在较低电流密度下运行,且催化效果不佳.此外,Co-NC-AF 与随机取向多孔氮掺杂钴单原子碳膜(Co-NC-RF)的明显析氢性能差别证明定向排列的微通道结构对于HER性能提升有重要作用.
2.2 析氧反应
通过太阳能电解水获得可再生能源(H2)和有价值副产物(O2)是利用太阳能的一种方式.然而,阳极缓慢的析氧反应(OER)动力学限制了电解水能量转化效率的提高,设计高效的单原子催化剂用于析氧反应是实现高效电解水的关键[46].在OER电化学反应条件下,催化剂表面大部分活性位点发生结构自重构,例如单原子的聚集,催化剂的催化性能可能发生改变[47].因此,发展高效OER单原子催化剂的一个主要挑战是稳定单原子位点.
Jiang等[22]通过电化学活化过程提前使催化剂表面自重构,前驱体np-Ir/NiFeP发生自重构,制备了稳定的铱单原子催化剂np-Ir/NiFeO.np-Ir/NiFeO在碱性电解液中展现出优于商用IrO2的OER性能[图7(A)~(C)]:在10 mA/cm2电流密度下,过电位为197 mV;Tafel 斜率为29.6 mV/dec;质量活性39.3 A/g;在1.43 V(相对于可逆氢电极)下进行计时电流测试,阳极电流80 h以内没有下降.依据原位X射线吸收光谱结果:当阳极电位进一步升高至1.55 V,相较于1.44 V时,X射线吸收近边结构(XANES)谱的白线强度增强[图7(D)],但傅里叶变换-扩展X 射线吸收精细结构(FT-EXAFS)谱没有变化[图7(E)],表明在较高电位下,铱价态的增加不是源于进一步的化学吸附或中间产物的形成[图7(F)].据此推测,在OER电化学反应过程中,铱原子的羟基配体去质子化导致中心原子氧化,价态升高,但配位环境不变,由铱原子位点的单一活性中心演变为铱原子位点与活性氧物种位点的多活性中心(Ir-O),加速了OER 反应过程.理论计算表明,反应的决速步是OOH*中间体的形成[图7(G)],而Ir-O结构单元更有利于OOH*中间体的形成[图7(H)],Ir-O位点可能是优异活性的来源.材料在电化学反应中表现出良好的稳定性,源于构建了稳固的表面结构.如图7(I)所示,Ni(Fe)—O键的收缩使得表面结构更加稳固,铱单原子被牢牢固定,防止了其团聚行为.此外,得益于双连续纳米孔结构的高效气体运输和基底大的表面积,在电压为1.48 V时,电流密度可高达300 mA/cm2.
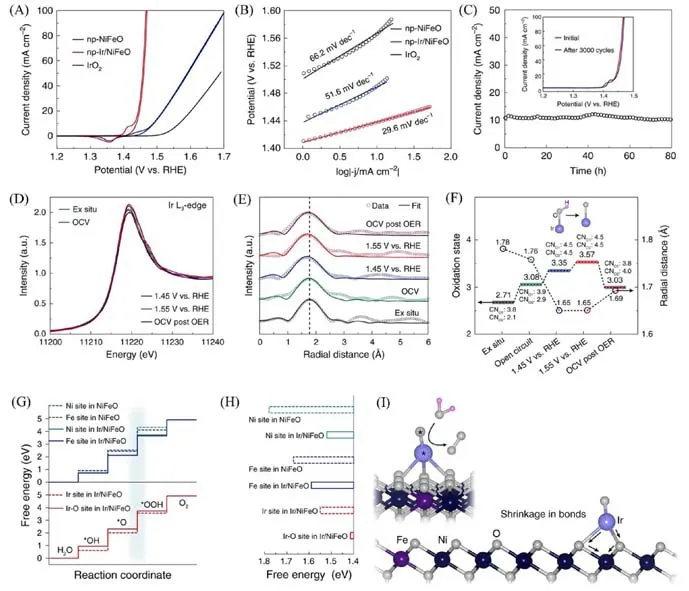
Fig.7 OER polarization curves of np⁃NiFeO,np⁃Ir/NiFeO and IrO2(A),corresponding Tafel plots(B) of the presented data in (A),stability of np⁃Ir/NiFeO evaluated by the current density versus time(i-t) curves at 1.43 V versus RHE with inset showing the CV curves of np⁃Ir/NiFeO before and after the accelera⁃tion durability test for 3000 cycles(C),operando XANES spectra of np⁃Ir/NiFeO recorded at Ir L3⁃edge under different applied voltages from OCV to 1.55 V versus RHE in 1.0 mol/L KOH(D),correspon⁃ding first⁃shell(Ir—O)fitting of FT⁃EXAFS spectra for np⁃Ir/NiFeO(E),the fitted oxidation states from the white line intensity analysis,the variation of Ir—O bond and the FT⁃EXAFS curve⁃fitting analysis(F),calculated free energy diagram of the OER(G),the Gibbs free energy of the rate⁃determining step for different sites(H),scheme for the shrinkage of the Ni(Fe)—O bonds determined by the operando XAS analysis of np⁃Ir/NiFeO(I)[22]
2.3 电化学制过氧化氢反应
在各种催化反应中,氧还原反应(ORR)是最重要的过程之一,众多能源储存系统与化工生产均涉及该过程.ORR过程包括多电子转移步骤,可通过4e−路径产生H2O(或OH−),2e−路径生成H2O2.在燃料电池等应用中,2e−ORR过程的发生意味着氧气的不完全还原,能量效率低且H2O2的生成会导致离子聚合物及膜的降解,更期望发生的是4e−ORR过程[48];而对于H2O2的生产,则更希望ORR过程趋于2e−路径进行,以实现高产率.因此,探索高效、耐用且选择性良好的ORR催化剂对于不同的实际应用具有重要意义.通过合理调控中心金属原子、配位原子、环境原子(碳基掺杂的异质原子)及外来基团,可以有效提高本征活性[48],其中中心金属原子的选择对催化剂的ORR性能具有最直接最显著的影响.对于4e−ORR,大多数研究认为,Fe是单原子催化剂最好的中心金属原子,Co稍差,但仍有较为出色的活性[48];对于2e−ORR,Co作为单原子材料中心金属原子,催化性能出色[49].
过氧化氢是一种非常重要的化学品,广泛应用于工业领域[50].蒽醌法仍是制备H2O2的主要方法,但耗能大,且产生污染物.电化学氧还原制备H2O2是一种有潜力的替代方式[51].许多研究已经报道了应用于ORR的单原子催化材料,但多为粉末材料,墨水形成的催化层的导电性和传质往往受到影响.基于此,Zhang等[52]合成了具有分级结构的自支撑钴单原子催化剂(Co SA/CC),在电压为0.1 V时,其电流密度达到51 mA/cm2,可进行连续10 h以上的电化学还原反应,选择性保持在80%.与Co SA/CC电极相比,由粉末状钴单原子材料制备的电极(金属量相同,涂覆于碳纸上,Co P/CC)在相同条件下的还原电流密度明显偏小[图8(A)],选择性也更差,这可能与部分催化位点被覆盖有关;阻抗谱[图8(B)]结果显示,Co SA/CC电极上的电荷转移电阻更小,电荷转移更快,展现其无需添加绝缘黏结剂的独特优势.将Co SA/CC 电极直接组装成电化学池生产H2O2,在外加电压为1.6 V 时,生产速率达到676
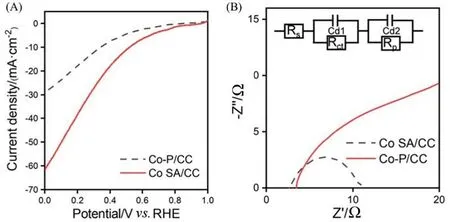
Fig.8 LSV curves in 0.5 mol/L H2SO4 at the scan rate of 10 mV/s(A) and Nyquist plots of Co SA/CC and Co⁃P/CC(B)[52]
2.4 锌空电池
由于柔性电池具有使电子产品更容易弯曲、适应性更强、使用更舒适的潜力,一直是人们感兴趣的研究对象.锌基电池安全系数高、能量密度大,在柔性电池领域备受关注[53].锌空电池的理论能量密度为1086 Wh/kg,具有成为可穿戴柔性电池的潜力.但空气电极缓慢的ORR/OER 动力学导致了锌空电池能量效率低、过电位大、稳定性差[54],绝缘且非活性黏结剂的使用更是加剧了这些问题[9,54].锌空电池理想的空气电极材料需要同时满足具备良好的ORR/OER 催化性能,且尽可能不使用黏结剂.同时,柔性电池需要以柔性为导向设计材料与系统[53],因此目前应用于锌空电池的自支撑单原子膜电极通常柔性较好.
Ji 等[28]设计了碳纳米纤维膜上生长负载有钴单原子的氮掺杂多孔碳片阵列(Co SA@NCF/CNF)材料,一体化的结构避免了黏结剂的使用.由图9(A)可见,Co SA@NCF/CNF相互连接的分级多孔阵列结构体现出极高的单原子利用率和快速的传质能力,并且碳纳米纤维膜保障了快速电子转移和机械性能,使其成为优异的ORR/OER双功能催化剂[图9(B)和(C)],ORR半波电位为0.83 V;在0.7 V电压下,24 h 内电流密度几乎没有变化;在电流密度为10 mA/cm2时,OER 过电位为400 mV.由图9(D)和(E)可见,组装的凝胶固态电解质可穿戴锌空电池具有优异的性能,开路电压为1.41 V;电流密度为6.25 mA/cm2时,比容量为530.17 mA·h/gZn;倍率性能优异,不同电流密度下电压下降幅度小;在循环900圈以后,性能没有明显变化,稳定性好.
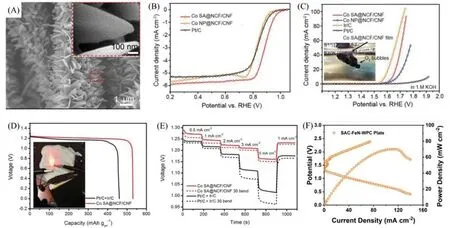
Fig.9 Field emission scanning electron microscope(FESEM) image of Co SA@NCF/CNF film(A),ORR LSV curves of Co SA@NCF/CNF,Co NP@NCF/CNF,and Pt/C in 0.1 mol/L KOH electrolyte(B),OER LSV curves of Co SA@NCF/CNF,Co NP@NCF/CNF,Ir/C,and Pt/C in 1.0 mol/L KOH electrolyte(C),the voltage⁃capacity curves of the prepared ZABs(D),discharge curves of the prepared ZABs at different current densities and testing conditions(E)[28],galvanostatic discharge/charge cycling curves and the corresponding power density of an SAC⁃FeN⁃WPC plate⁃based quasi⁃solid⁃state battery(F)[41]
Zhong等[41]以自支撑木质结构多孔碳负载的单原子材料(SAC-FeN-WPC)直接作为空气电极,组装可充电液态电解质锌空电池.稳固的自支撑结构、分级的孔隙结构、均一分散的Fe-N-C单原子位点使得SAC-FeN-WPC 的OER/ORR 性能出色,进而在锌空电池中展现出优异的性能.电池开路电压为1.53 V,充放电电压差、峰值功率密度均优于对比样(铂碳、氧化钌混合催化剂,Pt/C+RuO2),说明SAC-FeN-WPC具有更好的催化性能,且倍率性能、稳定性能优异.由图9(F)可见,在半固态锌空电池中,SAC-FeN-WPC 作为空气电极展现出70.2 mW/cm2的大功率密度.Qiu 等[55]以镍、氮共掺杂三维多孔石墨烯材料(Ni-N-np-G)作为自支撑空气电极,组装的全固态锌空电池最大功率密度也达到了83.8 mW/cm2.镍金属在材料中以单原子和团簇的形式存在,并且质量分数达到23%.Ni-N-np-G 是柔性极好的薄片状材料,因此即使锌空电池弯曲不同的角度,电池性能也不会受到影响,在柔性能源设备中具有应用前景.
2.5 二氧化碳还原反应
二氧化碳的过度排放造成了一系列的气候问题,将CO2转化为具有高附加价值的燃料或化学品是降低其在大气中含量的一条重要途径,通过电化学还原处理CO2拥有巨大潜力.CO2是非常稳定的分子,其进行电化学还原时需要催化剂活化.大多数CO2还原催化材料都在传统的H型电解池中测试评估,催化层完全浸泡于电解液中,CO2由液相电解液供应[30,56],但在电解液中CO2溶解度低、扩散缓慢,极大限制了反应速率[11].为此,使用包含气体扩散电极的反应器件(如流动池)受到关注,CO2变为借助气体扩散电极的气相供应,足量的CO2能够快速、短距离地到达催化剂表面,实现大电流条件下的电催化转化.但粉末状催化材料通常需要附着、沉积于气体扩散电极[57],电催化剂与基底之间的结合力较弱,容易脱落[58],聚合物黏结剂的使用也降低了电极的导电性.自支撑单原子材料则将气体扩散层与催化活性层整合为一个一体化结构,以其作为CO2还原催化剂克服了上述问题.
Zhao等[15]制备了两侧生长有N-CNTs的碳纸负载的镍单原子材料(H-CPs),在H型电解池中测试其将CO2还原生成CO 时的催化性能.在电压为−1.0 V 时,电流密度为48.66 mA/cm2,法拉第效率高达97%;在−0.7~−1.2 V范围内,选择性保持在90%以上.优异的性能可归因于镍单原子的高催化性能以及N-CNTs独特的垂直排列结构.N-CNTs的垂直排列结构提供了优异的导电性能,且其超亲水和超疏气表面有助于传质及气体产物脱离催化剂表面.研究特别讨论了材料的超亲水与超疏气性[图10(A)].H-CPs的N-CNTs阵列具有超亲水性,接触角接近0°;阵列的纳米粗糙结构减小了气泡与材料的附着力,在气泡体积不大时便脱离材料,体现出超疏气性.而与此相反,如[图10(B)]所示,氮掺杂碳负载的镍单原子催化剂附着于碳纤维纸(Ni SAs/CFPs)展现出较强的疏水性,且由于没有N-CNTs 阵列,气泡与材料附着力强,在体积较大时仍附着于材料上,当其脱离材料时必然带来很大的局部应力.值得一提的是,该催化材料制备简单,可规模化生产;自支撑材料直接作为工作电极,免去了粉末状材料制备工作电极的步骤,减少了相关成本投入.
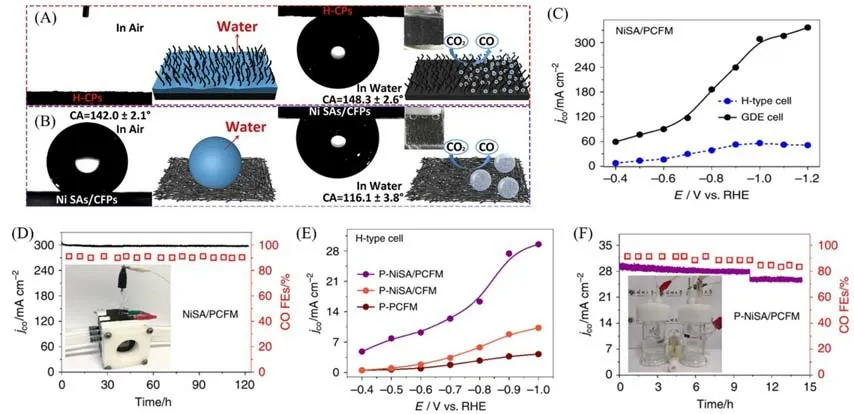
Fig.10 Water contact angles,underwater gas bubble contact angles and schematic illustration of H⁃CPs(A)and Ni SAs/CFPs(B)[15],partial current densities of NiSA/PCFM at various cathode potentials in GDE cell and H⁃type cell(C),stability tests of NiSA/PCFM in GDE cell at -1.0 VRHE(D),partial current densities for different catalysts at various cathode potentials in H⁃type cell(E)and stability tests of P⁃NiSA/PCFM in H⁃type cell at-1.0 VRHE(F)[30]
Yang等[30]合成了负载镍单原子的多孔(大孔、微孔)碳纤维膜(NiSA/PCFM),直接将其作为气体扩散电极,实现了在大电流工况条件下的CO2电催化转化.在配备空气扩散电极的流动池(GDE 池)中,当电压为−1.0 V时,生成CO的电流密度达到了308.4 mA/cm2[图10(C)],法拉第效率为88%,连续运行时间超过120 h(性能保持95%以上)[图10(D)].CO2还原发生在固-液-气三相界面,其在催化剂表面的扩散、吸附和产物脱离很大程度上决定了反应速率[59].不同于传统电极催化层的纳米级别厚度,NiSA/PCFM厚度在几百微米,电极不会被电解液完全浸没,从而有利于建立稳固的固-液-气三相界面;NiSA/PCFM 相互贯穿的多孔碳纳米纤维网络为CO2扩散、电子转移提供连续路径,分级多孔结构增强对CO2吸附能力,使得单原子周围富含CO2;催化剂特殊的多孔三维结构使镍单原子大部分都成为有效的催化活性位点,Ni-N4-C 的单原子配位结构在CO2转化为*COOH 中间体时,自由能更小,对于CO2电还原催化起着决定性作用;虽然NiSA/PCFM表面是亲水的,但经少许Nafion改性,可变为疏水性,对催化剂的长期稳定性有重要影响.基于以上原因,NiSA/PCFM 具有非常优异的催化性能.此外,研究中的一些对比实验也给出了有趣的结论:当电压为−1.0 V 时,NiSA/PCFM 在H型电解池中生成CO 的电流密度为56.1 mA/cm2,仅是其在GDE 池中的20%[图10(C)],体现出流动池装置的优势(提供充足CO2);在H 型池中,NiSA/PCFM 电极与涂覆于碳纸上的粉末状NiSA/PCFM(P-NiSA/PCFM)电极最高生成CO的法拉第效率相当,但前者的电流密度远高于后者[图10(C)和(E)],说明黏结剂的添加影响反应电流密度大小;不同于NiSA/PCFM 的良好稳定性[图10(D)],P-NiSA/PCFM 在H型池中于较小电流下,10 h后出现了明显的电流下降[图10(F)],原因可能在于催化材料的脱落.
Li等[60]通过酸活化、镍离子吸附及热解等简单步骤,在商用碳纸上植入镍单原子,合成了S、N掺杂碳纸负载的镍单原子催化剂(ACP/S-N-Ni).镍单原子与3个氮原子和1个硫原子配位,并且处于高氧化态(>+2),基于独特的配位结构,ACP/S-N-Ni电极具有较出色的CO2RR性能.将其应用于CO2电化学还原为CO的反应中,在660 mV过电位下,电流密度为3.4 mA/cm2,选择性达到91%,并且能连续工作至少14 h.由于商用碳纸较小的本征电化学活性面积,使得ACP/S-N-Ni的性能比不上最好的粉末状镍单原子催化剂,但ACP/S-N-Ni电极规避了催化材料在电化学反应中的脱落问题.更重要的是,该研究提供了一种将金属单原子位点植入碳纸或其它类型碳基基底的思路,以较简单的方式制备自支撑单原子电极应用于更多电化学反应.
2.6 锂硫电池
锂硫电池具有高理论比容量(1675 mA·h/g),且硫元素储量丰富、价格低廉,对环境友好,是非常有潜力的下一代储能系统.但锂硫电池目前面临着缓慢的反应动力学、穿梭效应导致的容量衰退、载硫量和硫利用率低等挑战,距离商业化应用还有很长一段距离.传统的涂覆法制备硫电极是将材料与活性物质、黏结剂、导电助剂制成浆料涂覆于集流体,但这种硫电极存在一些问题[8]:(1)材料涂层可能会与集流体脱离;(2)绝缘的黏结剂降低电极的导电性,同时降低电池的能量密度;(3)为保障电极的导电性,涂层通常较薄,载硫量较低.而自支撑电极材料无需添加黏结剂和导电剂,也无需集流体,其活性物载量往往较高[8,61~63],电池能量密度也更大[64].单原子催化剂常用来提升锂硫电池的性能,单原子位点的优异催化性能可加速活性物质转化的动力学,同时不饱和配位环境对多硫化物具有化学吸附作用.
Han等[65]制备了一系列负载于三维电极上的金属单原子催化剂(SAM-CFs,M=Mn,Cu,Cr,Ti),载硫量达到8 mg/cm2,其中SATi-CFs催化剂在充放电倍率为0.1C和3C时,面能量密度分别达到了8.2和5.1 mA·h/cm2,超过了商用锂离子电池的能量密度[图11(A)].由图11(B)可见,SATi-CF催化剂的催化性能优于多种过渡金属单原子催化剂.通过理论计算,发现低原子序数过渡金属具有更少的反键填充态和更有效的d-p轨道杂化(与硫物种之间),在多硫化物还原和硫化锂氧化过程中,能垒更低,揭示了SATi-CF催化剂拥有更好性能的原因.图11(C)所示硫化锂在电极表面的沉积行为证实钛单原子的作用:没有单原子的表面成核位点少,硫化锂以小尺寸颗粒的形式不均匀地分布于材料表面;与之相反,在SATi-CF表面,单原子可以作为活性位点引导选择性成核和生长,避免表面钝化,硫化锂沉积较厚、粗糙,体现出更强的硫化锂沉积能力.Huang等[61]通过静电纺丝技术合成了自支撑多孔碳纳米纤维膜负载的钴单原子催化剂[图11(D)],将其作为硫电极、锂电极的夹层组装成全电池,在6.9 mg/cm2的高硫载量下,其面能量密度达到7.15 mA·h/cm2(0.1C)[图11(E)].其出色的性能源于材料作为硫载体对活性物质转化的高效催化作用,以及作为锂电极侧夹层时,由于其亲锂特性,可以控制锂沉积行为,均匀化局部的电子/离子通量,抑制锂枝晶的产生.
理论上,只要自支撑电极的厚度(体积)增加,其载硫量就可以提升.但随着电极厚度的增加,电荷转移动力学变得缓慢,电极的传质受阻,活性物质的利用率降低,倍率性能更是难以令人满意.因此,提升自支撑电极导电性能,通过特殊的结构设计提高传质能力将有助于实现更高的载硫量和更大的能量密度.
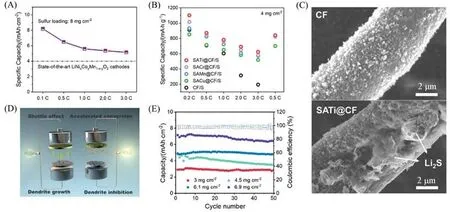
Fig.11 Comparison of the areal capacity of the SATi@CF/S cathode and commercial Li⁃ion batteries at diffe⁃rent rates(A),rate performance of different cathodes(B),SEM images of the final Li2S deposit on CF and SATi@CF surfaces(C)[65],schematic configuration of a conventional Li⁃S full battery and Co⁃PCNF dual⁃functional fibrous skeleton enabled Li⁃S full battery(D),areal capacity of S/Co⁃PCNF||Co⁃PC⁃NF@Li full batteries obtained at 0.1C with different sulfur loadings(E)[61]
3 总结与展望
本文综述了自支撑单原子膜电极的研究进展,包括材料的合成策略及在能源电催化中的应用.自支撑单原子膜电极的合成策略总体上分为在已有自支撑基底上原位合成单原子及在合成自支撑骨架的同时引入单原子,如静电纺丝法、自组装法、化学气相沉积法、固相扩散法等.自支撑单原子材料在HER、OER、CO2RR、锌空电池、锂硫电池等反应及器件中已被广泛应用,表现出优异的催化性能.
与粉末状浆料涂层电极相比,自支撑单原子膜电极材料在大电流密度电催化转化、涉及气体传输扩散的电化学反应过程、以及高能量高功率密度电池等应用中具有独特优势,具体体现在以下几个方面:(1)导电性增强.整体式电极结构规避了绝缘黏结剂的使用,形成一体化的导电骨架,有利于电子的转移传输.(2)机械性能增强.不同于粉末状材料依靠黏结剂较弱的黏附力形成催化层,自支撑单原子膜电极各部分“无缝衔接”,电极结构更稳固.对于涉及气体的电化学反应,稳固的电极结构可抵御气泡带来的局部应力.(3)活性位点利用率高.以自支撑方式构建的材料界面,单原子在基底上原位生长,活性单元与基底的相互作用增强,活性位点结构被高度限制于基底.在增强活性位点稳定性的同时,可有效避免单原子位点被包埋,暴露更多活性位点.(4)形貌、结构、表面性质易调控.依赖于自支撑单原子膜电极形貌、结构的可调控性,具有分级多孔结构、特殊微通道结构、亲(疏)水界面等特点的膜电极均可制备.孔隙结构带来的大表面积造就了大量可参与电化学反应的单原子位点,增加了电化学活性面积;特殊的微通道和孔隙结构保障了电极的传质能力(电解液、气体的迁移扩散),进而实现大电流电催化反应的目标.自支撑膜电极通常可直接作为气体扩散层与催化层“合二为一”的空气扩散电极.(5)活性材料占比高.凭借无需添加黏结剂和导电助剂的优势,整个自支撑膜电极均是活性材料,在同等条件下,活性位点数量更多.将其作为电池电极,可实现高能量高功率密度储能系统的构建.总体而言,自支撑结构工程有助于提高电催化性能.
尽管面向能源电催化应用的自支撑单原子催化材料的相关研究已经取得了一些进展,但仍然有许多问题需要解决.(1)自支撑材料的物理性质、化学性质、几何结构等与电催化性能之间的构效关系需进一步明晰,这将为材料的理性设计与合成提供理论指导.该过程需要使用更多表征技术,从多角度的性能指标综合分析.(2)电化学性能的改进对于自支撑单原子材料的发展至关重要.除了开发具有更复杂的分级几何结构、孔隙结构,以引入更大的活性表面积,暴露更多单原子活性位点,提高传质能力外,应该评价不同金属源的催化性能,增加单原子载量.同时材料表面的亲/疏水性也应在合成材料时综合考量.(3)结构、机械稳定性对于长期循环使用至关重要.催化活性部分与支撑基底之间的连接必须稳固,需要发展更多合适的技术、方法来进一步增强单原子与基底的连接作用;基底材料本身的机械性能也需要通过优化成分配比和结构设计来增强.(4)自支撑材料的厚度决定了活性位点的数量和作为电池电极时活性物质的载量.对比粉末状材料涂层的厚度,自支撑材料的厚度已有明显增加,但制备电化学性能不受厚度影响的自支撑电极材料仍需付出更多努力.(5)自支撑单原子材料的实用化仍面临挑战.自支撑单原子催化剂的制备成本较高,降低生产成本、实现大规模、省时地制备自支撑材料是走向实际应用的必要条件;实现材料多次利用的技术手段欠缺,因此,催化剂的回收与再利用技术也值得重视与发展;缺乏相应实际应用组件的设计、优化,应当根据自支撑单原子材料的特点为其设计合适的反应器件,以充分发挥其催化作用.(6)自支撑材料的性能评价体系有待进一步完善,还需对标工业化生产要求来确定可靠的测试标准.在大电流条件下,传统粉末材料已不能作为一个标准来衡量自支撑材料的电化学性能;自支撑材料在电解液环境中进行稳定性测试时,其机械稳定性的评估标准也应统一化.
