石墨炔原子催化剂的崭新道路:基于自验证机器学习方法的筛选策略
2022-06-29黄汉浩卢湫阳孙明子黄勃龙
黄汉浩,卢湫阳,孙明子,黄勃龙
(香港理工大学生物及化学科技学系,香港 999077)
当前,现代社会急速发展伴随的能源需求的急剧增加已成为制约科技发展和社会进步的重要问题之一,因此可再生能源对于人类的长远发展起着至关重要的作用.目前,通过电催化方式生产的氢气、氧气及氨气等常用的清洁能源是可再生能源的重要组成部分,已逐渐受到了大量关注[1~7].高效稳定的电催化剂能够持续地将自然能源转化为可用能源[8~10].其中,过渡金属(TM)具有多样化的氧化还原能力,通过耦合或掺杂可满足不同催化条件的需求.此外,通过合金化或表面应变进行表面电子修饰等处理方式制备纳米电催化剂也是制备过渡金属基电催化剂的常用方式[11~18].目前,降低制备成本并同时提高其耐用性是电催化剂领域的关键问题.尽管非均相过渡金属基电催化剂[如铂(Pt)、钯(Pd)和铜(Cu)基电催化剂],在水分解[19,20]及二氧化碳还原反应(CO2RR)[21,22]中均展现了良好的催化效力,但其造价高昂.同时,传统的非均相电催化剂只有表面原子参与反应,在金属原子利用率方面受材料固有局限性的限制.相比之下,均相催化剂通常原子利用率更高,但稳定性较差,容易发生团簇,影响催化效果.
单原子催化剂(SACs)结合了均相催化剂和非均相催化剂的优势[23,24]且具有极高的原子利用效率,在催化效率和其它性能方面都表现出巨大优势[25~29].SACs的特点是活性位点独立,分工明确,催化效率极高,根据该特性可制备高选择性和催化活性的电催化剂[30].但热力学稳定性较低的性质导致SACs容易因为高表面能而发生团簇.在经历一系列表面反应的过程中,单个金属原子难以保持长时间处于一个固定位置,因此,需要找到一种适宜的衬底材料,能够在最大载荷的前提下保证单金属原子的稳定性,同时抑制团簇的发生.碳材料储量丰富,具有大量的同素异形体,而且廉价,化学稳定性高,生态友好,因此是材料领域的热门研究主体之一[31~33].在碳的同素异形体中,石墨炔(GDY)得益于其出色的电化学特性,是理想的单原子催化剂的衬底材料.石墨炔是一种二维结构的碳同素异形体,由sp和sp2轨道杂化的碳原子构成,苯环部分由丁二烯键连接,形成十八碳六方环[34].GDY独特的孔洞结构能够提供丰富的空间给金属原子,降低空间结构引起的金属原子不稳定性.此外,石墨炔由炔键和苯环连接而成,特别是其富炔碳骨架具备独特的sp和sp2结合的电子结构,使其具有高度的π共轭和可调电子结构,能够与负载的金属原子稳定成键,从而很好地俘获及稳定金属原子,最终实现均匀的金属原子负载,并有效提升催化剂的活性面积,降低金属载量.而且GDY具有高比表面积和炔键等结构特征,为GDY功能化提供了大量的反应位点.与传统的二维碳材料(例如石墨)相比,石墨炔具有天然的带隙,这归因于其2pz轨道的重叠以及sp2和sp杂化的碳原子之间不均匀的π键[35~37].石墨炔独特的高电子电导率和载流子迁移率等特性能够实现高效催化,因此可以作为一种有效锚定金属原子的衬底结构[38~40].例如,利用石墨炔锚定稳定的过渡金属元素(镍和铁)可以实现零价金属原子高效催化水分解产氢(HER)[41].
尽管SACs已被证明是高活性高效率的催化剂,但实际应用中仍面临着单原子结构带来的一些问题.SACs 在反应中间物的吸附能之间存在着难以打破的线性关联(Scaling relation),在实际应用中不可避免地限制了催化效率[42~44].例如一氧化碳还原反应产生的废气中含有大量的碳氢化合物和氮氧化物,在使用SACs进行反应时,各类反应物的竞争吸附会极大降低反应效率[45],而由多原子构成的原子催化剂(ACs)是解决该问题的有效手段.譬如引入一个新的原子构成双原子催化剂(DACs),引入的新原子会提供更为灵活的催化活性位点,使多种反应物同时吸附,同时优化结合键能,促进中间态转化,诱导协同效应,从而显著提升催化性能[46~51].最近,关于石墨炔-双原子催化剂的报道中提到,即使有石墨炔结构的衬底,依旧难以架构稳定的双原子催化剂系统.金属原子固有的热力学稳定性特征与最佳组合方式的选择难度导致制备石墨炔-双原子催化剂困难重重.此外,制备SACs和DACs通常选择过渡金属,对于主族元素一般少有关注[52].目前普遍认为除过渡金属之外的元素能够有效抑制团簇聚集,因为它们通常有较高的聚集能势垒,使得原子迁移的概率大大降低.传统的实验-试错过程在面对大量的潜在组合方式时需要耗费大量的时间和经济成本,这使得计算技术在促进石墨炔原子催化剂(GDY-ACs)的设计合成上拥有巨大的优势.通过计算技术可以实现实验预设计、合成指导和预测结果等关键步骤.本文综合评述了近期的一些关于GDY-ACs电催化剂的工作进展,通过一系列的密度泛函理论(DFT)与机器学习算法的研究策略证明了不同金属组成的GDY-ACs 在促进可持续能源发展方面的潜力,同时,本课题组的研究也表明机器学习算法能够有效推动GDY-ACs的合理设计,为未来设计先进催化剂提供了重要思路.
1 筛选稳定的石墨炔-原子催化剂
目前的研究认为,作为催化剂的金属原子与作为衬底的其它原子之间的相互交互作用决定了表面反应的活性和催化剂系统的稳定性.一些研究也证明了石墨炔与金属原子之间存在较强的电荷迁移,因此是的理想衬底材料[53~57].同时,理论算法和机器学习等先进设计方式进一步促进了石墨炔-原子催化剂组成方式的发展.与传统的单原子催化剂相比,零价金属原子催化剂突破性地表现出了更高的稳定性与催化活性.但零价金属原子催化剂的优越性不能简单归因于活性位点的价态,对于活性位点电子结构的修饰才是性能增强的主要成因[41].零价态是金属原子-载体相互作用引起的电荷补偿的结果,石墨炔的电学特性使得被锚定的金属原子和衬底结构之间存在大量的电子迁移,形成了具有零价态的石墨炔-原子催化剂系统.
1.1 筛选零价原子催化剂
为了寻找性质更加优良的零价催化剂,本课题组通过计算方式筛选了大量过渡金属-石墨炔的组合.通过理论计算手段筛查各组合的电荷转移效率和稳定性,在所有过渡金属(IIIB至IIB)的范围内,研究了潜在的理想催化剂材料及将其锚定在石墨炔表面的可行性(图1)[58].被锚定的单金属原子通过电荷补偿维持在稳定的零价态,这是由过渡金属-石墨炔体系良好的电子转移特性决定的.我们用氧化还原反应的能量势垒来解释过渡金属-石墨炔体系的电荷转移能力:还原反应能垒越高,原子越倾向于保持在零价态;相反,氧化反应的能垒较高则表明金属原子更倾向于处于高价态,并且难以回到零价态.通过DFT 计算方式得出的过渡金属从3d到5d轨道的氧化还原能的结果显示,当3d-过渡金属处于+2价的初始状态时,由于还原反应能垒阻碍较大,还原反应占主导地位,大多数3d-过渡金属表现出较高的零价态稳定性[图1(A)].而考虑到4d轨道时,Pd的4d轨道具有较高的还原反应能垒,也适用于石墨炔基零价单原子催化剂[图1(B)][59].出乎意料的是,计算得出的Tc和Cd的氧化还原能垒也较高.在所有4d-过渡金属中,只有Rh主要由氧化反应主导氧化还原趋势,表明Rh难以形成稳定的零价原子催化剂.5d-过渡金属整体的锚定稳定性略低于3d-和4d-过渡金属[图1(C)],但Hg表现出绝佳的零价稳定性,而其它常用于催化剂的金属,如Ir和Re也表现出非常强的稳定零价趋势.另外,常用的贵金属催化剂如Au,Ag和Cu,计算结果表明其难以在锚定状态下保持零价态,但最近Li等[53]成功地将零价铜原子锚定在石墨炔上用于催化水分解产氢,表明计算方式的结果仍存在局限性.另一方面,氧化还原能垒也用于评估催化反应性.过渡金属原子成功锚定后大大降低了石墨炔的氧化还原能垒,表明锚定金属原子后增强了电子迁移效率,使得石墨炔的催化活性大幅提升.基于能量的筛选原则表明,d6-d9电子结构的元素氧化还原能垒高于1 eV,表明这类材料在锚定石墨炔后会表现出高度的零价稳定性,是具有潜力的稳定催化剂材料[图1(D)].除Ti和Pd之外的其它电子结构(约0.75 eV)则显示出较低的零价稳定性.与+2价态的情况相比,通过得电子形成+1价态和零价的能垒都大于2 eV,表明发生电子迁移的难度较大.从电子亲和力的角度也证明了零价态稳定性,3d到5d过渡金属的电子亲和力随电子结构呈现波动趋势,而在3d-过渡金属中,Co和Ni表现出了高电子亲和力,这与其在氧化还原能垒中表达的高零价稳定性一致.Pd在4d-过渡金属组中表现出了最高的电子亲和力.在所有过渡金属中,Os显示出了最高的电子亲和力,而Ti和Ir的低电子亲和力可能是由于氧化还原能垒较低,因此难以保留电子.另一方面,计算结果表明,Ln通常表现出稳定的非零价态,这意味着它很难如过渡金属原子一样保持稳定的零价态.总体而言,Pd,Co,Pt和Hg是实现稳定零价过渡金属-石墨炔催化剂的优势候选组合,它们在大量电子迁移时表达了强烈的还原趋势,同时能够在初始电荷迁移时保持稳定的零价态[图1(E)~(I)].通过空间最高占据分子轨道(HOMO)和最低未占分子轨道(LUMO)分布以及模拟电子能量损失近边结构(ELNES)计算图示,GDY-Pd,GDY-Co和GDY-Ir等催化剂组合结构中出现明显的电荷密度重新分布,这是催化水分解产氢时快速进行电荷交换的先决条件[57].除了对于各类金属元素的本征特征及合成结构的基础属性的计算之外,本课题组也提出了先进的深度学习算法和大数据技术在合理设计以及进一步修饰复杂的原子催化剂方面的优势,使用模糊C-均值(FCM)算法,以核函数为筛选函数,可实现基于数据聚类的数据集分类.
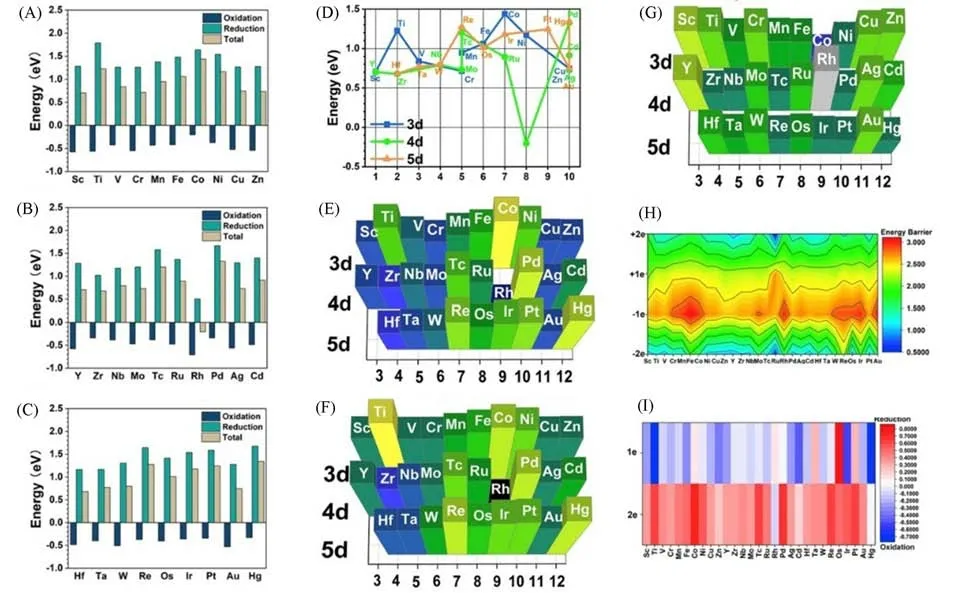
Fig.1 Redox energy diagrams,zero⁃valence anchoring ability and mapping of the electron transfer ability of metal on GDY[58]
1.2 筛选双原子催化剂
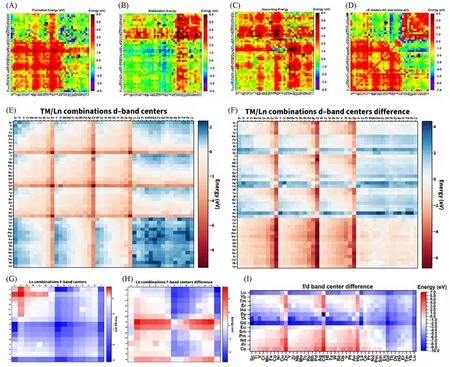
Fig.2 Mapping of formation energy,stabilization energy,anchoring energy and d,f⁃band center energy of GDY⁃based DACs[60]
除石墨炔基单原子催化剂之外,计算方法也可用于寻找稳定的石墨炔-双原子催化剂(图2)[60].实验合成良好的石墨炔-双原子催化剂面临两个关键问题:制备过程中的热力学稳定性和难以确定的最佳原子组合.需要注意的是,即使本身过渡金属原子与石墨炔之间的存在强相互作用,也不意味着能够成功地合成石墨炔-双原子催化剂,因为如果形成过渡金属原子二聚体在能量角度并非最优选择,则两个过渡金属原子可能会分别形成单独的单原子催化剂而不是双原子催化剂[图2(A)~(C)].d-d轨道之间的强排斥力大大降低了均耦合(TM1-TM1)和异耦合(TM1-TM2)石墨炔-双原子催化剂的热力学稳定性,导致难以合成石墨炔-过渡金属双原子催化剂[图2(D)].研究表明,石墨炔-过渡金属双原子催化剂的形成能遵循3d-3d<4d-4d<5d-5d的规律,而3d-4d,3d-5d和4d-5d过渡金属原子组合的形成能差距不大.这些结果都表明过渡金属原子极为倾向形成孤立的原子位点,这是合成石墨炔-单原子催化剂的优势,却是合成石墨炔-双原子催化剂的劣势.由于镧系元素的f轨道与过渡金属原子的d轨道能够形成强烈的耦合效应,它们组合可以显著降低双原子催化剂的形成能.对于Ln-3d组合来说,绝大部分的搭配都具备热力学稳定性.该组合优异的稳定性和高电活性来源于Ln-TM原子之间长程位点f-d轨道相互作用,通过f-d轨道耦合作用可以实现Ln-TM DACs系统中的电子自平衡效应.同时,与3d-3d和5d-5d同源耦合的双原子催化剂相比,Ln-Ln同源耦合双原子催化剂在热力学稳定性上更为突出.其中Sc-Pr,Y-Pr,Au-Sm和Nd-Sm组合是Ln-3d,Ln-4d,Ln-5d和Ln-Ln组合系列中最为稳定的双原子催化剂组合.然而,即便有良好的热力学稳定性,基于镧系金属原子的双原子催化剂相比于镧系单原子催化剂选择性更低,因此难以确保双原子催化剂的合成.在电活性方面,之前的研究表明,d带的位置与电活性密切相关[61~63].通常,电催化剂中靠近费米能级的d带中心能保证在反应过程中进行快速的电子迁移.对于基于过渡金属的GDY-DACs,伴随着d带上的电子增加,整体d带位置呈现向下迁移的趋势.从3d-到5d-过渡金属,3d-(3d,4d,5d)GDY-DACs 的整体d带中心呈下降趋势,而4d-(3d,4d,5d)和5d-(3d,4d,5d)GDY-DACs也同样表现出类似的下降趋势.基于过渡金属的双原子催化剂整体d带位置偏低表明,即使与d带中心较高的过渡金属原子耦合,基于双过渡金属的GDY-DACs仍然表现出较差的电子迁移能力,证明d-d轨道之间的电子迁移并不能带来高效的电活性反应.对于基于镧系金属原子的GDY-DACs,计算结果显示具有两个峰的驼峰状d带中心趋势,中间部分的镧系金属原子d带中心位置处于“峰谷”位置,电活性较低.双镧系金属原子合成的GDY-DACs 由于d轨道完全占据,因此d带中心穿过费米能级,表明镧系双原子催化剂处于高价态.同时,过渡金属-镧系金属原子的组合能够拉高d带中心位置,表明镧系元素的f电子对于双原子催化剂价态的调制作用[图2(E)和(F)].Ln-TM DACs提供了更丰富的电子结构,并且能够促进电催化过程中的电子迁移,同时自电子平衡效应也缓解了d带中心过低导致的催化活性不足.Ln-Ln DACs 的f带中心则显示出明显的谷状趋势[图2(G)和(H)],表明镧系元素中轻与重的镧系金属原子具有高电活性.总的来说,最稳定的Ln-Ln组合通常也牺牲了其作为双原子催化剂的电活性.相比之下,由于自平衡效应,Ln-TM组合显示出更好的热力学稳定性和更为适合电催化的电子结构[图2(I)].计算结果表明,Ln-Ni,Ln-Rh,Ln-Rh,Ln-Pd,Ln-Os,Ln-Ir和Ln-Pt等双原子催化剂组合都具有适合的d带中心位置(在费米能级1.0 eV内)以适应各种电催化过程.值得注意的是,经测算,Co-Nd,Ni-Er,Ru-Pr,Rh-Pr,Pd-Nd,Os-Pr,Ir-Pr 和Pt-Ho的配合方式在考虑形成能的前提下不仅能够实现稳定合成,同时具有高电活性.此外,通过高斯过程回归(GPR)算法能够验证DFT测算的结果.使用金属原子的基本物理化学性质对算法进行进行预训练,GPR算法表明GDY-DACs的热力学稳定性更依赖于锚定金属的基本参数,而电子性质,如d带中心和电活性则更受金属/金属和金属/衬底轨道耦合的影响.
除了TM和Ln原子,主族元素原子也能有效催化热反应[51,64~67].与过渡金属原子相比,主族元素,包括碱/碱土金属(AAEM)、后过渡金属(Post-TM)和类金属元素(Metalloid)原子,都很少用于电催化剂.然而,Post-TM和类金属元素原子的团簇能垒相对较高,因此极有可能形成稳定的原子催化剂[68].此外,将主族元素与TM 和Ln 金属原子耦合能够制备高电活性的稳定GDY-DACs(图3)[69].最重要的是,我们的工作还证明了p-d/f轨道相互作用在GDY-DACs催化过程中扮演着关键角色[69].对于AAEM原子,它的价电子主要位于s轨道,这导致在与TM原子耦合时s-d轨道相互作用较弱,使得过渡金属原子位点的电子结构和电活性变化都十分有限.AAEM-Ln GDY-DACs的形成能是所有组合中最低的,因为s轨道的强屏蔽效应导致与f轨道的过度耦合[图3(A)~(D)].由于从AAEM到TM的原子s轨道对局部电场产生强烈扰动,导致整体能量趋势缺乏规律性.就Post-TM而言,即使轨道取向限制了Post-TM和GDY之间的p-d耦合,占主导作用的p轨道也可以通过有效的p-p轨道耦合赋予GDY烷基链相对较高的电活性.出于对稳定性的考虑,Post-TM GDY-DACs 具有比基于碱/碱土金属元素原子的GDY-DACs更高的整体形成能.Post-TM-TM 和Ln 金属原子之间的强排斥力导致这两类金属原子合成的GDYDACs的双原子之间经常长距分离.而对于类金属原子,由于s轨道的屏蔽效应被抑制,占主导地位的sp轨道杂化会引起交换关联效应.由于过度结合效应,类金属原子可以作为分隔介质来稳定单个分散的过渡金属原子,并且通过与其它的过渡金属原子形成稳定的原子二聚体来避免GDY-DACs 发生团簇.对于基于类金属-镧系金属元素原子的GDY-DACs,它们表现出相对较弱的耦合,形成能接近零线(0 eV).对于基于镧系金属元素原子的GDY-DACs,s/p-f轨道耦合和排斥力同时存在,但大多数基于Ln金属原子的GDY-DACs从能量角度来看仍然表现良好,因为大多数被锚定的金属原子之间的排斥力相对较弱.但Ln金属原子对AAEM和Post-TM金属元素表现出很强的排斥力.总体而言,我们的计算筛查表明TM-TM,Post-TM-TM 以及Post-TM-类金属这3 种不同的GDY-DACs 组合形成能较高,难以大量稳定合成.另一方面,基于AAEM元素原子的双原子催化剂比单原子催化剂的形式更稳定,而类金属原子在和TM元素原子合成双原子催化剂时能够起到稳定作用.这些结果证明了存在使用AAEM和类金属元素原子作为“分隔介质”的可能性.大多数基于碱金属元素原子和Post-TM或类金属元素原子组合的DACs热力学稳定性良好,但基于碱土金属元素原子和Post-TM或类金属元素原子组合的DACs热力学稳定性则要差得多.同时,主族元素原子和Ln 金属原子的结合可以制备极为高效稳定的GDYDACs.总体而言,用Ln元素原子合成的GDY-DACs具有最稳定的结构,尤其是在与AAEM元素原子配合使用时.我们的筛选计算还表明,较低的热力学稳定性通常来源于高度敏感的p和d轨道,而s和f轨道强有力的屏蔽效应有利于削弱被锚定的原子之间的排斥力,从而形成高度稳定的电催化剂.由于和AAEM、Post-TM 和类金属元素原子配合合成的非均相过渡金属GDY-DACs 的形成能相对更低[图3(E)~(G)],因此除了Nb-Nb和Hf-Hf这两类GDY-DACs之外,基本不采取合成均相过渡金属GDYDACs的方案.而从电子结构角度[图3(H)~(J),图4][69],AAEM元素原子的p带中心即使与其它元素耦合也难以产生明显的变化,表明AAEM在石墨炔-双原子催化剂中相对惰性.然而,由于引入了碱性离子,烷基链的p带被推向费米能级附近,表明碱性离子的s轨道电子能够激活烷基链的电活性来促进电子迁移.这种对于烷基链的修饰影响也可以通过添加Post-TM元素原子达到,但由于被锚定原子之间的强相互作用,Post-TM元素原子产生的调制作用十分微弱.而类金属元素中的Se原子也同样可以活化烷基链,从而改善电子迁移效率并实现高效的电催化.再考虑到过渡金属和镧系金属原子的d带中心或f带中心,锚定AAEM元素原子会造成4d-或5d-过渡金属原子的d带中心下降,而Post-TM金属元素原子对d/f带中心几乎无法产生影响,仅有Al和Pb能够显著激活Pt-5d和Au-5d这两种组合,并且通过降低能垒提升电子迁移效率.Post-TM 拥有高度的p-d和p-f轨道耦合,能够有效分隔TM/Ln SACs的活性位点并同时保留了高度的电活性.此外,计算结果显示基于类金属原子的GDY-DACsd/f带中心高度收敛.总体而言,我们的研究表明,添加AAEM、Post-TM 和类金属元素原子不会对已锚定的TM和Ln金属元素原子的电活性产生显著影响.此外,我们也通过输入元素的固有特性,包括质量数、重量、电负性、电子配置、p/d/f带中心、电离能、电子亲和力、原子半径和两个锚定原子的电导率等,对GPR算法进行反复训练与适应,并使用训练后的GPR算法对GDY-DACs的能量趋势和电子结构进行评估.结果表明,当p轨道作为影响因素之一被引入计算系统时,通过固有性质训练后的算法系统无法完全估算形成能.而引入p轨道带来的影响也证实了计算s区元素将会有更高的难度.总体而言,对电子结构的预测精度在不同元素组中差异显著,基本顺序为Post-TM(p区)>类金属(s,p杂化)>AAEM(s区),AAEM 的p带中心预测显示出较大偏差,均方根误差(RMSE)达到了2.185,其原因在于p带中心的高度不连续兼或变化范围极大.而除AAEM参与之外的其它组合的预测结果则与DFT数据的偏差较小,尤其是Post-TM GDY-DACs的组合.这表明数据偏差主要来自于基于AAEM的GDY-DACs的p带中心的不连续区域,这可能是由被锚定原子之间的轨道耦合引起的.
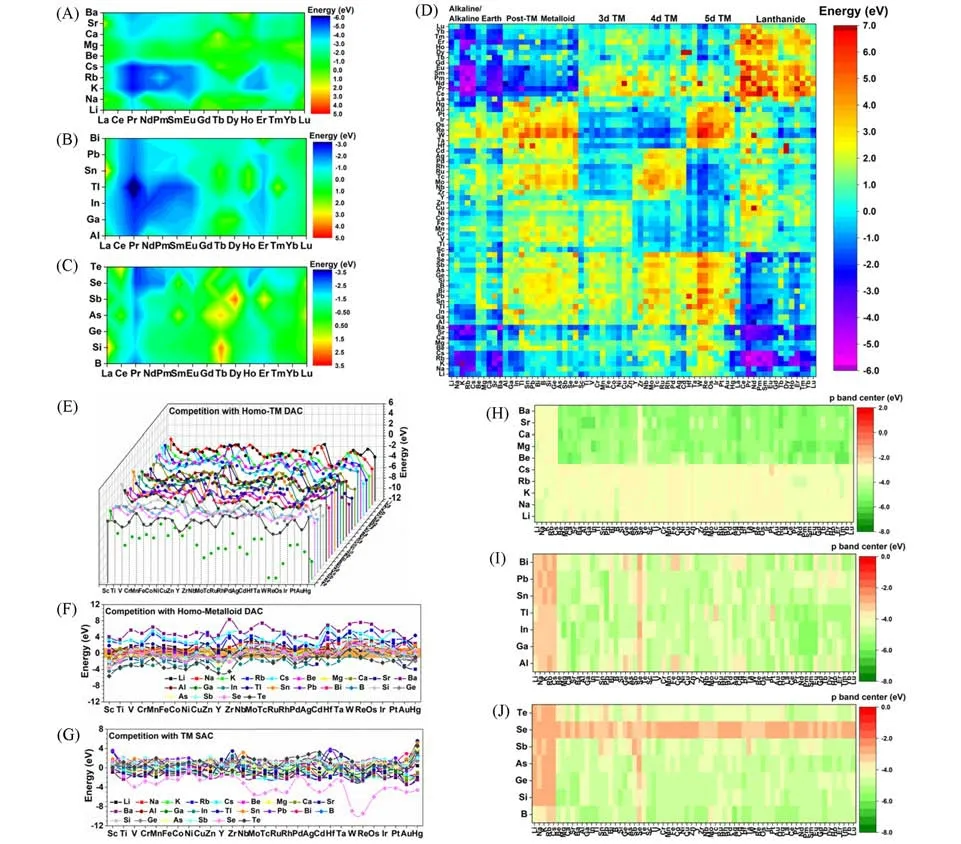
Fig.3 Formation energies and band center mapping of GDY⁃based DACs[69]
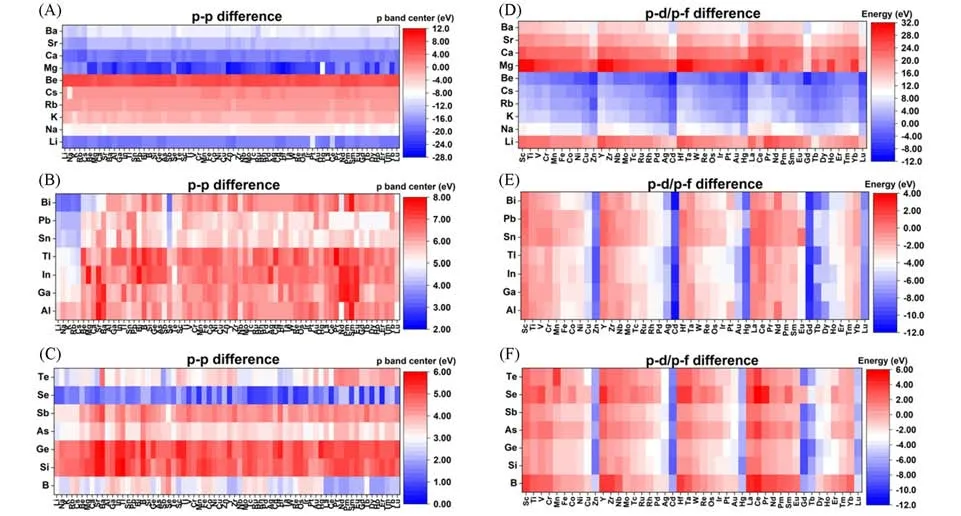
Fig.4 Orbital coupling of GDY⁃based DACs[69]
2 石墨炔-原子催化剂催化水分解产氢
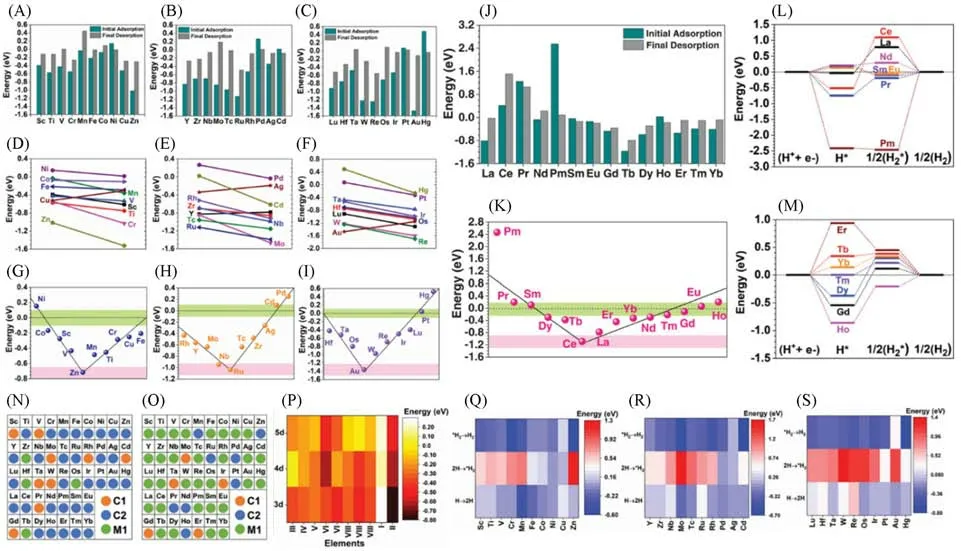
Fig.5 Adsorption of TMs based GDY ACs and their HER performances[70]
基于前述部分研究,本课题组从吸附能角度出发进一步研究了GDY-ACs 催化水分解产氢的过程(图5)[70].传统的计算方式通常将氢原子的吸附能作为最重要的评价参数来衡量水分解产氢的表现,而我们的工作则将理论计算与机器学习技术相结合,筛选出适用于HER 的GDY-ACs 组合.包括Volmer,Heyrovsky 和Tafel 等关键步骤在内的反应过程都对HER 性能产生着至关重要的影响,而评估HER过程中的整体能量变化和趋势则可以通过比较H2脱吸附能和H吸附能来完成[图5(A)~(C)].对于3d-TMs,大多数金属都表现出比初始H吸附能更高的H2脱吸附能,并且两者差值较小,但Mn和Zn的两者差值较大,在催化HER过程中拥有极大优势.另外,Ni是3d-TMs中仅有的表现出自发反应趋势的元素,证明其催化HER的整体效应良好.4d-过渡金属原子的初始H吸附能与H2脱吸附能之间的差值则表现出了更大的变化,其中仅有Pd和Cd表现出了适宜催化HER的性能.Pd的最终H2脱吸附能接近0 eV,能够有效产生氢气[59].除了Pt和Hg,大多数5d-TM并不适用于GDY-ACs,因为往往需要外加极大的电势差来克服在HER中的吸热效应.其中Pt的整体反应趋势高效有利,并且最终H2脱吸附能极低,而Hg催化HER的效率则因为初始H吸附阶段大量吸热而受到极大影响.氢的化学吸附趋势可以作为衡量氢覆盖的指标参数[图5(D)~(F)].通过能量角度估算,大多数过渡金属元素在氢的化学吸附阶段能起到良好的作用,这表明了持久的氢吸附能力是HER 中Heyrovsky 反应的关键基础.镍基GDY-ACs的氢吸附趋势自发并且稳定,因此可以避免催化剂中毒.而GDY-Zn,GDY-Mo和GDY-Ru等系统表现出了对氢的强结合趋势,这种趋势极容易导致过度结合,使得H2的脱吸附能大大提升.值得一提的是,尽管GDY-Au表现出了极强的氢吸附能力,但是其化学吸附趋势却并不具备优势.Pt和Hg的氢化学吸附趋势都很强,这进一步证实了这两类金属元素原子在高效催化HER方面的潜力.此外,通过GDY-AC的初始吸附和最终脱吸附之间的能量差,我们进一步评估了TM-GDY在氢吸附上的表现[图5(G)~(I)].而Zn 位于火山图(Volcano plot)的谷点位置,表明其催化HER 的能力最弱.从初始吸附和最终脱吸附之间的能量差判定,Ni,Fe 和Co 是3d-TMs 中催化HER 表现最佳的候选材料.在4d-TMs中,Cd和Pd的能量差最小,而Ru位于中间谷点,还原能力较弱,难以实际催化HER.尽管化学吸附计算表明Ru基GDY-AC并不能有效催化HER,但Li等[71]的研究表明,他们合成的Ru-GDY-AC是高效的水分解电催化剂,在酸性条件下对HER和OER都具有高效稳定的催化活性,强有力的p-d耦合使Ru成为一种独特的电子介导载体(Electron-mediating-vehicle),可以实现可逆的氧化还原快速切换.在5d-TMs 中,Au 位于最低点,只有Pt 显示出较高的HER 催化潜力,因为其能量差几乎为0 eV.总体而言,4d和5d金属都表现出了更大的能量差值(超过0.4 eV),表明与3d金属相比,它们催化HER 的效率较低.对于基于Ln的GDY-ACs[图5(J)~(M)],大多数Ln金属在初始吸附和最终脱吸附之间的能量变化处于一个中等范围.然而,广泛使用的电催化剂Ce由于其强大的氧化能力而显示出较差的HER催化潜力,Pm也由于其初始阶段氢吸附需要消耗极高能量而不适用于催化HER.而Sm,Eu,Gd和Ho等镧系金属在H2生成过程中整体呈现自反应趋势,表明它们是适宜催化HER的可能选择.除Pm,La和Ce之外的镧系元素的能量差值都处于一个固定范围内,其中Sm,Gd和Eu的位置最接近中线.总体而言,GDY-Eu和GDY-Sm这两种是已被证明的能够有效催化HER的电催化剂,经过计算可得,它们在吸附第二个氢原子和脱吸附H2阶段的能量消耗较小,这与实验结果相符.此外,通过基于不同轨道电子态密度(PDOS)的电子结构研究,我们进一步验证了GDY-ACs 的电子迁移能力.在3d金属中,Cr,Ni,Co和Fe的3d主峰都位于费米能级附近,表明它们具有良好的电子迁移效率.而d轨道被完全填充的过渡金属元素,如Cu和Zn等,由于其深埋的3d轨道,电子迁移本应受限,但GDY-Cu仍能凭借石墨炔优秀的电荷迁移能力高效催化HER[53].通过观察4d-过渡金属的整体趋势,可见其4d主峰逐渐迁移,而从Mo到Rh这一系列元素的t2g和eg轨道之间存在明显带隙,造成了不利于催化HER的能垒.Pd则由于4d峰靠近费米能级附近而显示出极高的电活性,这不仅能表明Pd 具有优异的催化HER 的能力,同时也是GDY-Pd 能够有效催化氮还原的理论证明[72].Ag 和Cd 表现出与Cu 和Zn 相似的电子结构,说明它们并不适合作为电催化剂.而在5d-过渡金属中,Os,Ir 和Ptd轨道峰值与费米能级接近,因此有催化HER的可能性,其中Pt-GDY已经实验证明[56].从电子结构的角度,我们评估计算了各类金属GDY-ACs的电活性[图5(N)~(S)],其规律遵循3d>4d>5d,这个结论与前述的通过能量差值得到的结论相吻合.此外,由于Eu和Sm的4f轨道具有良好的电子迁移能力,GDY-Eu和GDY-Sm也表现出极高的电活性.在考虑对GDY-ACs上的活性位点进行筛选时,我们的研究表明,由于单个金属原子易与氢原子产生过度吸附效应,因此金属原子位置不是理想的稳定吸附位点.另一方面,C1对H2的吸附最不稳定,而GDY 链上的C2是包括Co和Pt在内的所有GDY-ACs上最具电活性的位点[图5(N)和(O)].为了验证通过DFT 计算得到的结果,我们使用袋装树方法(Bagged-tree method)作为机器学习算法,通过引入大量原始数据训练系统来预测GDY-ACs的吸附能,其中包括理论计算结果、电负性、电子亲和力、电离势、d/f电子、活性位点和质量数.随着引入氧化还原能垒作为计算的新参数,对于质子的化学吸附能的预测精度得到了显著提高,表明质子的化学吸附能与氧化还原能垒密切相关,由元素的内在特征决定,而不是受外在的非电子交换的物理吸附的影响.通过比较DFT计算与机器学习得出的质子的化学吸附能,表明机器学习技术的精度较高.但机器学习算法的结果也仅能重现整体能趋势,元素之间的详细差异仍需要进一步改进算法.
3 总结与展望
本文综合评述了在GDY-ACs设计优化方面的最新研究进展.这些工作表明,充分结合理论计算和机器学习技术在提升先进有效的GDY-ACs 的设计方式和进一步提升性能方面发挥着关键的作用.通过制定筛选策略,证明了Co和Pt作为稳定的零价过渡金属GDY-ACs的潜力.同时,Ln-TM DACs组合也被证明是极具潜力的电催化剂候选组合,因为该组合能够通过长距点对点的f-d轨道相互作用实现电子自平衡.此外,我们也探索了基于主族元素的GDY-DACs,结果显示主族元素与TM 和Ln 金属的结合可以形成具有极高热力学稳定性的GDY-DACs,长程p轨道耦合的特质使得该类GDY-DACs 具有高电活性.DFT 计算也揭示了主族元素的p轨道的作用,表明它们对活化烷基链环节具有重要作用.而机器学习算法则进一步证实了s轨道和p轨道的不同作用.此外,DFT计算和机器学习技术也被用于评估GDY-ACs催化HER的能力,开创性地预测了包括稀土材料在内的可用于催化HER的石墨炔-原子催化剂组合的特征.这些研究还表明,GPR算法的不确定性量化能力揭示了选择训练数据内容产生的巨大影响,这可以加深我们对催化剂参数与其性能之间关联性的理解.尽管我们的研究表明结合DFT计算和机器学习技术在预测包括热力学稳定性和电活性等各种催化剂性能方面拥有极大优势,但机器学习技术仍需要预先获取大量数据对算法进行训练以获得更为准确的预测结果.因此,开发需要较少数据的算法方式将会大大减少研究人员对资源的需求.为了开发一个可以覆盖更广泛受众的算法模型,与计算机科学家进行更密切的跨学科合作至关重要.同时,我们在热力学稳定性和催化HER电活性方面的理论预测还需要进一步的优化去支持实验结果,这表明GDY-ACs的热力学稳定性和催化性能在实际应用中并不仅受简单的几个因素的控制,需要考虑通过构建更复杂的模型使理论预测更接近现实,未来在原子催化剂领域的发展还需要更多先进理论研究策略的支持与指导.
除了本文探讨的HER,石墨炔原子催化剂在未来不同的催化反应中也具有很大的潜力.与传统碳材料相比,石墨炔在合成及结构方面均具备独特的优势.首先,石墨炔的制备条件温和,不需要高温合成,在100 ℃以下的液相体系中能够在多种基底上实现原位大面积制备,这突破了一般传统碳材料需要高温高压等条件的制备方式.此外,石墨炔具有天然均匀的平面多孔结构,通过前驱体的设计可以进一步实现多孔尺寸的调控,这是一般碳材料所难以实现的.而且石墨炔是所有碳材料中唯一具有sp和sp2杂化的二维碳结构网络的材料,兼具独特的催化活性和物理稳定性.此外,石墨炔的能带结构和力学性质等也可以通过不同的杂原子和官能团修饰进行调控,可以在未来针对不同的催化反应进行精准设计.目前,石墨炔原子催化剂已经在多个不同的催化领域实现了应用,包括水分解产氧(OER)、水分解产氢(HER)、氮还原(NRR)等不同电催化反应.而目前在氧还原(ORR)领域,石墨炔原子催化剂的发展还有待进一步开拓.目前研究已经发现氢原子修饰的石墨炔能够在碱性条件下表现出优于商业碳载铂催化剂的超高活性,同时具有比Pt/C更好的稳定性和抗甲醇中毒能力,具备在燃料电池中应用的潜力.此外,相较于一般商用Pt/C,石墨炔原子催化剂的应用能够大幅降低燃料电池的应用成本,这些优势与前期研究都为未来石墨炔原子催化剂在多样化能源系统(如燃料电池)中的应用奠定了基础.
