单原子催化剂在锂硫电池中的研究进展
2022-06-29尹肖菊赵程浩赵晨阳张乃庆
尹肖菊,孙 逊,赵程浩,姜 波,赵晨阳,张乃庆
(1.哈尔滨工业大学交叉科学研究中心,2.化工与化学学院,哈尔滨 150008)
锂硫电池以单质硫为正极、金属锂为负极,具有高的理论能量密度(2600 W·h·kg−1)[1,2],同时,硫具有储量丰富、价格低廉、环境友好及易于回收等优点.因此锂硫电池成为高能量密度电化学储能领域的重要发展方向[3,4].与锂离子电池脱-嵌锂储能机制不同,锂硫二次电池主要通过单质硫与金属锂之间的氧化还原反应实现化学能与电能间的转化,其充放电反应过程较复杂,涉及多步、多电子转移和多相转变过程[5,6].锂硫电池尽管在能量密度上和其它二次电池相比具有很大的优势,但仍存在活性物质硫利用率低、循环寿命短及倍率性能差等问题,成为其进一步广泛应用亟需解决的关键问题[7~9].
2009年,Nazar 等[10]采用高度有序的介孔碳CMK-3作为锂硫电池正极载体,将硫限制在介孔孔道内,通过物理限域束缚活性物质硫,电池表现出高放电容量和稳定的循环性能.从此,锂硫电池进入了一个新的发展阶段.随后,研究者们开发了大量多孔碳/硫复合体系[11~13].但非极性碳与极性多硫化锂(LiPSs)之间的相互作用比较弱,碳材料抑制LiPSs 穿梭的能力有限.考虑到化学亲和作用的重要性,电负性高的杂原子(如氮、硫、氟等)被掺杂到碳材料中,使其能通过化学吸附作用更有效地锚定LiPSs,减少穿梭效应[14~16].另外,进一步的研究发现,极性化合物(如金属氧化物、硫化物、氮化物、碳化物、磷化物等)除了锚定LiPSs外,还能作为催化剂提升反应动力学,在抑制穿梭效应的同时提高电池的倍率性能[17~21].
单原子催化剂(SACs)理论上具有100%的原子利用率,在不同的化学反应中表现出了非凡的催化活性[22~24].SACs的概念一经提出便引起了众多领域研究人员的广泛关注,成为近年来催化领域的研究热点[25~29],SACs在锂硫电池中的应用也得到了深入的研究[30~35].由图1可见,SACs能够应用于锂硫电池的正极、负极以及隔膜/中间层.SACs在应用于锂硫电池正极中时,其活性中心与LiPSs具有强的相互作用,可以用于锚定LiPSs;此外,由于具有极高的催化活性,其还可以通过加速LiPSs的氧化还原反应反应动力学、缩短LiPSs的存在时间来降低LiPSs 扩散的可能,抑制穿梭效应,提高活性物质利用率.SACs还可以用于修饰金属锂负极来保护负极,抑制锂枝晶的产生.高度分散的SACs 活性位点可以均匀调控锂的沉积,其亲锂特性有助于降低锂与沉积基底界面的能量,从而降低成核过电位,并抑制锂枝晶的形成.隔膜作为电池的重要组成部分,其作用是导通离子并防止短路,然而商业化聚丙烯(PP)隔膜由于孔径较大,多硫化物容易通过.单原子修饰的多功能隔膜不仅具有催化剂的作用,可以在正极侧抑制穿梭效应,还可以通过均匀调节锂离子通量有效防止锂枝晶的产生.
本文综合评述了SACs在锂硫电池中的正极、负极、隔膜/中间层的最新研究进展,并对SACs在锂硫电池中的未来发展方向进行了展望.
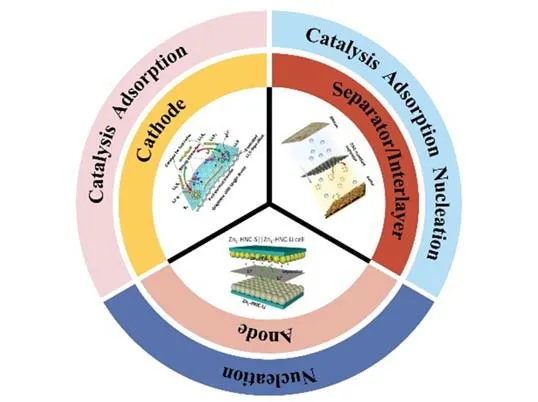
Fig.1 Schematic diagram of SACs in Li⁃S battery
1 SACs在锂硫电池正极中的应用
硫正极作为锂硫电池最重要的组成部分之一,具有高达1675 mA·h·g−1的理论放电比容量.但硫正极的研究面临以下几个突出问题:(1)放电中间产物LiPSs(Li2Sn,4≤n≤8)易溶于醚类电解液,引起穿梭效应,影响其循环性能;(2)LiPSs 转化过程动力学迟缓,电池的倍率性能差.针对以上问题,研究者相继提出了物理限域、化学吸附及催化转化等策略,其中催化转化策略为同时抑制穿梭效应和提升倍率性能提供了切实可行的解决方案.单原子催化材料由于具有独特的结构和高活性,近年来人们对其在锂硫电池正极中的应用开展了大量的研究.
1.1 不同金属中心SACs
负载在高导电性网络的SACs具有高的硫负载能力和电子导电能力及强的化学吸附作用,可锚定LiPSs,限制LiPSs 扩散;同时具有高的催化活性,可加速LiPSs 的转化过程,抑制穿梭效应,提高电池的循环性能和倍率性能.2018年,Yang等[36]以铁酞菁为铁前驱体、邻二苯胺为氮源、硅溶胶为硬模板制备了负载在氮掺杂的多孔碳材料上的单原子铁催化剂(Fe-PNC),并将其应用到了锂硫电池中.Wan等[33]报道了在氮掺杂石墨烯上负载的钴SACs(Co-N/G)并用于锂硫电池正极,通过球差校正扫描透射电子显微镜的高角度环形暗场成像技术(HAADF-STEM)观察到了Co 在石墨烯表面的原子分散状态,并且通过X射线吸收光谱的X射线吸收近边结构(XANES)和扩展X射线吸收精细结构(EXAFS)进行分析和拟合,给出了Co单原子的形态和配位构型(图2),证实了Co单原子是以Co-N4的配位构型嵌入石墨烯晶格中的.通过实验结合理论计算发现,单原子钴可以加速硫化锂的成核和分解过程,制备的硫正极S@Co-N/G在1C倍率下循环500次后,库仑效率仍高达99.6%.在6.0 mg/cm2的高S负载量下,在0.2C 倍率下循环100 次后的面容量为5.1 mA·h·cm−2,每次循环容量衰减率仅为0.029%.此后,通过不同方法制备的石墨烯以及介孔碳等碳基材料负载的多种SACs也被用于锂硫电池正极并且表现出了优异的电化学性能[27,35,37].
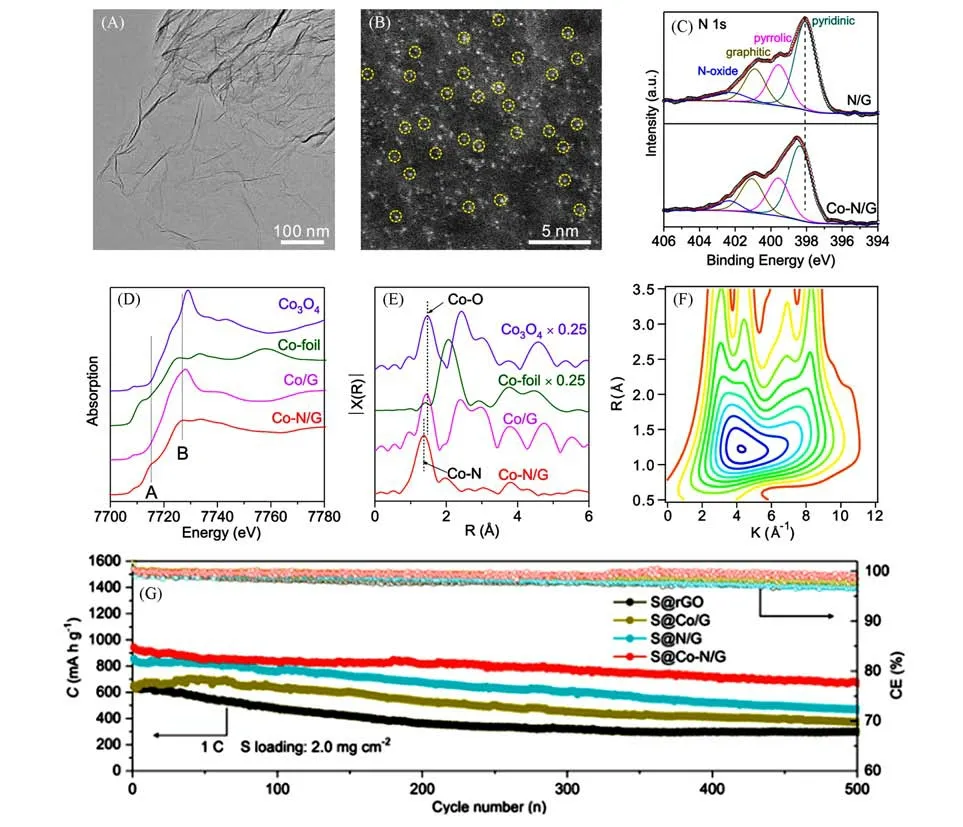
Fig.2 Structure and cycle perfomance of the Co⁃N/G composite[33]
理论计算是从电子结构和分子轨道角度研究LiPSs与吸附催化材料之间相互作用的有效工具,基于密度泛函理论(DFT)的第一性原理计算在阐释反应机制和提供设计策略方面具有独特的优势,能够在原子水平解释催化剂促进锂硫电池转化的反应机理.与实验试错法相比,理论计算被更系统、更广泛地用于预测和筛选过渡金属作为SACs.2020年,Cui等[38]以石墨烯、氮掺杂石墨烯以及氮掺杂石墨烯负载的铁、锰、钌、锌、钴、铜、钒和银SACs 等10 种材料作为研究对象,借助DFT 计算Li2S 在这10种基底上的分解势垒、锂离子扩散及其与LiPSs的相互作用强弱来综合分析筛选SACs,计算结果如图3所示.由图3可见,钒单原子(SAV@NG)表现出最低的分解势垒(1.10 eV).他们在理论指导下成功合成了氮掺杂石墨烯负载的钒SACs(SAV@NG)并应用到锂硫电池正极中,发现电池的放电容量、倍率和循环性能均得到了显著提高.
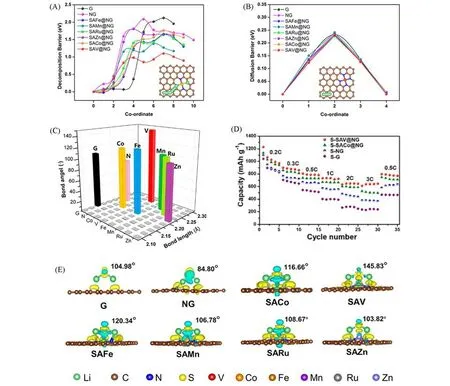
Fig.3 Theoretical understanding for Li2S decomposition,Li ion diffusion,and anchoring effect[38]
Cheng等[39]通过电子结构计算发现SACs和硫物种之间发生了d-p轨道杂化.其中,低原子序数的过渡金属,如Ti,具有较少的填充反键态和更有效的d-p轨道杂化,可降低LiPSs 还原和Li2S氧化过程中的能垒.制备的TiSACs 在8 mg/cm2的硫负载量下仍可以展现出8.2 mA·h·cm-2的高面容量.Han等[40]利用DFT研究了嵌入氮掺杂缺陷石墨烯(M@N/G,包括M@N3/G和M@N4/G)中的含3d,4d和5d电子的过渡金属SACs作为锂硫电池催化剂材料的性能,根据催化剂的稳定性、对硫物种的吸附自由能、从Li2S4到Li2S的自由能变化及Li2S的分解能垒等4个评价标准对M@N/G的性能进行评估,预测了负载在N4/G上的Sc,Cr,Mn,Ru,Os和Ir单金属原子催化剂具有良好的性能.
1.2 金属中心配位环境对催化性能的影响
除了改变金属单原子种类外,通过改变单原子周围中心原子的配位环境(如配位原子与配位数等)也会影响单原子的局域电子结构,改变其催化活性.Liu等[41]在氧、氮双掺杂空心碳球框架上负载锰单原子Mn/C-(N,O),通过XANES和EXAFS分析证明,O和N原子作为Mn原子的配位原子可以进一步稳定Mn原子,通过有效调节Mn原子的d电子密度提高Mn原子的催化活性,增强Mn原子的路易斯酸碱性,促进与LiPSs的化学相互作用.
Wang等[42]制备了单分散的铁氮掺杂多孔石墨烯(HFeNG),研究发现位于石墨烯层表面和空穴边缘的铁原子分别与4个氮原子(Fe-N4)或2个氮原子(Fe-N2部分)配位.DFT计算结果表明Fe-N2具有比Fe-N4更高的吸附能力,与多硫化物的相互作用更强.
Li 等[43]利用金属有机框架化合物(MOF)ZIF-8的自模板作用,制备了在中空氮掺杂多孔碳上负载的单原子Ni 材料(Ni-N5/HNPC).理论计算结果表明,与Ni-N3/C 和Ni-N4/C 相比,不对称电子分布的Ni-N5/C活性中心显示更低的势垒以及对LiPSs中等的吸附强度(图4).Chen等[44]还研究了过饱和Fe-N5配位结构的SACs,也得出了上述相似的研究结论.与M-N4结构相比,过饱和的M-N5结构具有更强的吸附能力.以吸附Li2S6为例,Fe-N5-C 对Li2S6的吸附能为-1.63 eV,而饱和配位的Fe-N4-C 的吸附能仅为-0.73 eV.以Fe-N5-C作为宿主材料所制备的硫正极展示出了优异的电化学性能,在0.2C倍率条件下具有1224 mA·h·g−1的放电比容量,即使在8.2 mg/cm2的高硫负载量以及4.7 μL/mg 的低电解质/硫(E/S)比条件下循环50次后,仍能保留高达6 mA·h·cm−2的面容量.
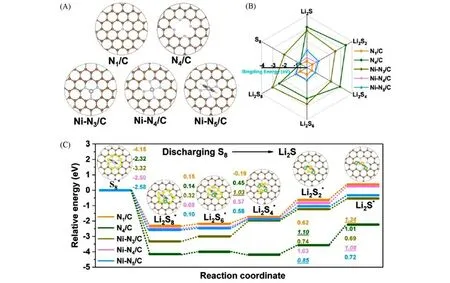
Fig.4 Computational design of Ni⁃N5/C[43]
1.3 催化剂载体的影响
中空纳米结构的碳在用作硫宿主材料时具有明显的优势,有足够的空间去容纳单质硫,缓解电极在充放电过程的体积膨胀.Wang等[45]通过Fe-ZIF-8的碳衍生物制备了固定在富氮空心碳笼的铁单原子(FeSA-CN)催化剂,催化剂中单原子铁负载量为1.14%(质量分数).DFT 理论计算结果表明,FeSA-CN中FeN4的正电性中心可以与LiPSs中负电性的Sx−偶联,大大降低Li+和Sx-之间的结合能,同时FeSA-CN 的正电性活性中心对LiPSs 中的Li+也有一定的排斥作用,Li+容易从LiPSs 中分离出来,所以FeSA催化位点可以通过以上两种途径降低Li2S的分解能垒.Chen等[46]在氮掺杂的介孔空心碳球上通过分解酞菁铁前驱体得到了单个铁原子嵌入的氮掺杂介孔空心碳球(Fe-N/MHCS).由路易斯酸碱相互作用产生的同时具有“亲锂”和“亲硫”位点的Fe-N4位点对LiPSs具有更强的化学吸附.制备的S@Fe-N/MHCS 电极在1.0C 倍率条件下的初始放电比容量为834 mA·h·g−1,循环1000 次后放电容量保持率为82.9%,容量衰减低至每圈0.0187%,同时库仑效率接近100%.利用该电极制备的S@Fe-N/MHCS软包电池在0.2C倍率条件下初始放电容量高达459 mA·h,对应于1257 mA·h·cm−2的放电比容量,循环200次后容量保持率高达77.1%.
除了非极性的碳材料外,使用极性材料作为单原子载体可以进一步增强SACs对多硫化物的锚定能力,抑制穿梭效应的发生.并且如何提高单原子的负载量也是SACs所面临的一个重要课题.Zhang等[47]以极性的氮化碳为载体,构建了具有Fe-N2不饱和配位结构的铁SACs(Fe-N2/CN),其中,铁原子的负载量高达11.4%(质量分数).以Fe-N2/CN为宿主材料制备的复合正极表现出了优异的放电比容量及循环性能(图5),在2C倍率下充放电循环2000次后,每次平均容量衰减率仅为0.011%;此外,当硫面负载量达到5.6 mg/cm2时,仍能表现出5.4 mA·h·cm−2的面容量.Yang 等[48]通过熔融氯化锌蚀刻Ti3AlC2(MAX)的方法在Mxene 上负载单原子锌材料(SA-Zn-Mxene),以SA-Zn-Mxene 作为硫宿主材料,它不仅对多硫化物具有优越的亲和力,而且能显著降低Li2S2到Li2S限速步骤的转化能垒.此外,SACs的金属载体强相互作用会导致单原子中心和载体表面的电荷重新分布,对活性中心的电子结构及催化活性产生显著影响[49,50].
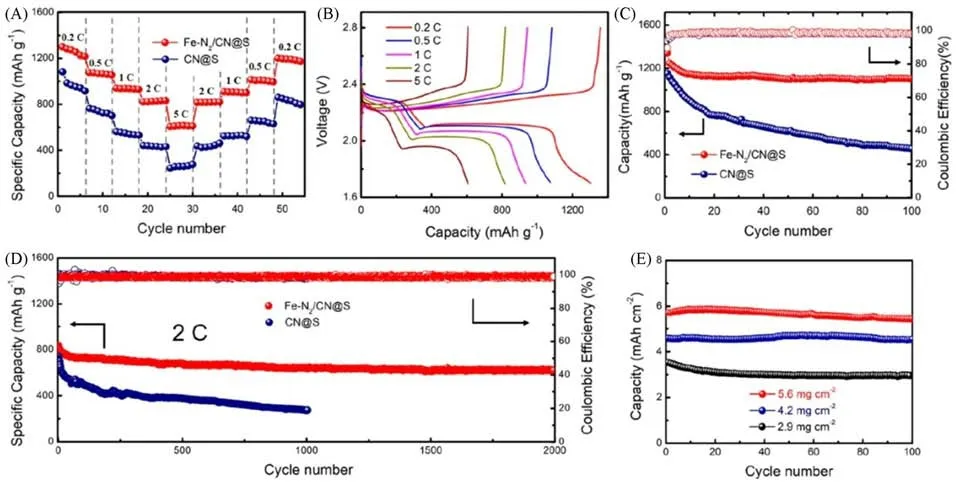
Fig.5 Electrochemical performance of Li⁃S batteries based on Fe⁃N2/CN@S and CN@S cathodes[47]
1.4 单原子和纳米粒子协同催化
随着单原子研究工作的深入开展,研究人员发现SACs对于多硫化物的吸附能力受到催化剂金属载量的限制.然而金属含量的提高往往会导致金属原子发生聚集,形成金属纳米颗粒或者金属化合物,无法实现单分散.因此,除了提高单原子载量的方法以外,与其它金属或金属化合物纳米粒子复合也是一种切实有效的提高催化剂性能的解决方案,并且二者之间会产生协同作用,进一步加速充放电过程的反应动力学,提高锂硫电池正极的电化学性能.Zhao等[51]认为SACs的活性位点为不能完全消除LiPSs的穿梭效应的单端结合位点,提出了嵌入极性硫化锌纳米粒子和Co-N-C SAC组合成双端结合位点的思路,并制备了三维有序大孔导电框架材料上同时负载极性硫化锌纳米粒子和Co-N-C的SAC材料(3D-omsh/ZnS,Co-N-C/S).Wang等[52]制备了一种由Co纳米粒子和Co-Nx共掺杂的碳纳米管固定在碳泡沫中的三维独立框架.作为一种新型的无黏结剂的硫宿主材料,这种新型正极在7.0 mg/cm2的高负载量以及5 μL/mg的低E/S比条件下,仍然展示出优异的放电性能和循环寿命.
2 SACs在锂硫电池负极中的研究进展
金属锂负极存在沉积/剥离库仑效率低和易生长锂枝晶等突出问题.单原子材料由于具有高密度的亲锂位点,能够诱导金属锂的均匀成核,有效抑制锂枝晶的生长.
Sun等[53]在静电纺丝法制备纳米纤维的溶液中添加(Co,Zn)配位的沸石咪唑骨架(Co-ZIF-8),然后热分解得到单原子分散Co-Nx的纤维碳骨架(Co-PCNF),并将其作为S正极和Li负极的载体(S/Co-PCNF和Co-PCNF@Li)[图6(A)].孤立的钴原子可以通过氮配位被限制在ZIF-8 衍生的载体中.由图6(B)可见,这种网络结构赋予纤维骨架优异的弯曲和折叠的机械性能.Co-PCNF应用于硫正极时能够有效地促进硫的电化学双向转化动力学,从而抑制了LiPSs 的穿梭效应;应用于锂负极时能够增强纤维状碳骨架的亲锂性,促进负极均匀的锂成核和生长,从而抑制锂枝晶形成.S/Co-PCNF||Co-PCNF@Li 全电池在0.2C 倍率条件下,硫负载量为3.5 mg/cm2时的循环寿命达到600 次,每次循环容量衰减率仅0.082%,即使在6.9 mA·h·cm−2的高硫负载量下,在0.1C 倍率下也能实现7.15 mA·h·cm−2的面容量.
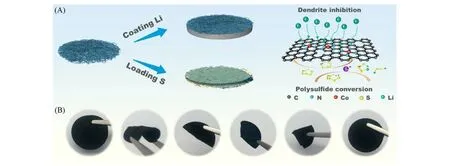
Fig.6 Functional illustration of the fibrous skeleton Co⁃PCNF[53]
Wu等[54]通过热解中空结构的ZIF-8前驱体合成了锌单原子改性中空多孔碳壳(Zn1-HNC)纳米反应器,单原子负载量高达15.7%.该纳米反应器可以同时作为硫正极(Zn1-HNC-S)和锂负极(Zn1-HNC-Li)的载体(图7).理论计算和实验证明Zn1-HNC 纳米反应器不仅可将多硫化物物理限制在空心壳内,而且对多硫化物的液-液转化反应具有高催化活性,从而实现锂硫电池的高硫负载和更好的循环稳定性.同时,Zn1-HNC 具有均匀的单原子Zn 亲锂位点,可有效抑制锂枝晶的生长.Zn1-HNC-S 正极和Zn1-HNC-Li负极组装的锂硫全电池在700次循环中仅有0.015%的容量衰减率,在10C倍率下仍然具有989 mA·h·g−1的放电容量.另外,该电池还实现了低E/S(6.4 μL/mg)和正负极容量比(1.8∶1).
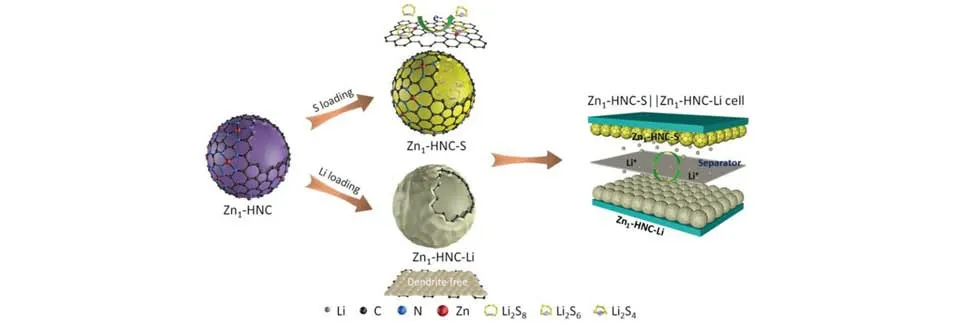
Fig.7 Synthesis schematic of the dual⁃functional Zn1⁃HNC nanoreactors for Zn1⁃HNC⁃S cathode and the Zn1⁃HNC⁃Li anode of the Zn1⁃HNC⁃S||Zn1⁃HNC⁃Li full battery[54]
3 SACs在锂硫电池隔膜/中间层中的研究进展
SACs 材料不仅能够作为锂硫电池的正极材料用于锚定多硫化物并促进多硫化物的转化,还能够用作电池的隔膜(或者修饰隔膜)和中间层,提升电池的电化学性能.由于隔膜和中间层非常薄,能够降低SACs 的用量,从而提高其利用效率.同时,SACs 中间层或其改性的隔膜能够有效地锚定多硫化物,并加速多硫化物转化的氧化还原动力学.
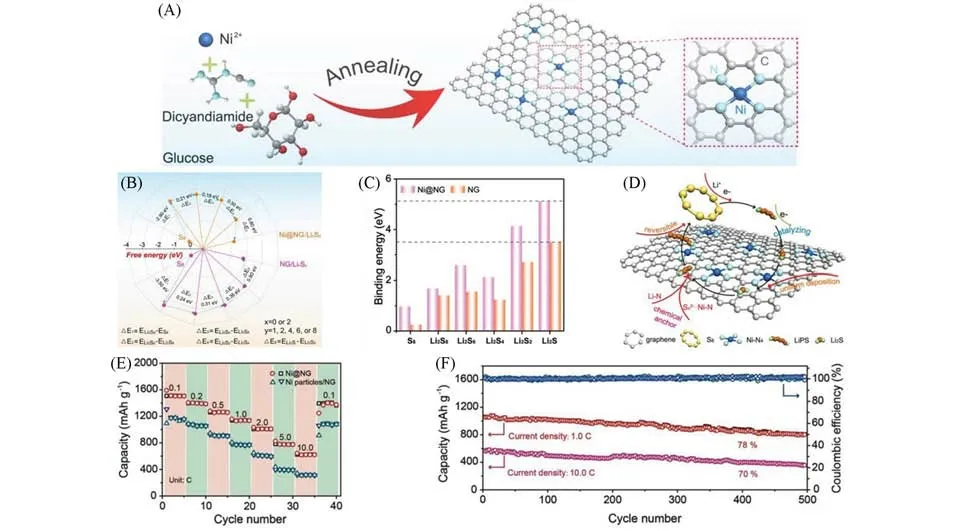
Fig.8 Schematic illustration and electrochemical performance of Ni@NG[34]
Niu等[34]参照文献[55]方法,以葡萄糖、双氰胺、NiCl2·6H2O为反应物,借助原位热分解的方法成功制备了负载在氮掺杂石墨烯上的Ni 单原子材料(Ni@NG,图8),并将其用于修饰锂硫电池的隔膜.Ni@NG独有的Ni-N4特殊结构使得负载的Ni原子能作为多硫化物的活性作用位点,通过形成化学键有效固定多硫化物,从而减缓其在电化学反应中的穿梭效应.此外,Ni@NG改性隔膜与电解质具有较大的接触角,其疏水特性的表面有利于通过表面排斥阻止极性多硫化物的穿梭.同时,Ni@NG对多硫化物的转化具有优异的催化活性,Ni原子可与多硫化物之间发生电荷转移,加快多硫化物的氧化还原反应,提高活性物质的利用率,显著提升电池循环寿命.利用Ni@NG 改性隔膜组装的电池在1C 倍率下循环500次后仍能表现出826.2 mA·h·g−1的可逆容量,容量保持率高达78%.Liang 等[56]将NiCl2·6H2O替换为FeCl3·6H2O,采用相同的原位热分解方法制备了负载在氮掺杂石墨烯上的Fe单原子高活性催化剂材料(Fe@NG),并将其用于锂硫电池的隔膜改性,取得了类似的结果.
Xie等[57]通过将氧化石墨烯在金属盐溶液(FeCl3,CoCl2或NiCl2)中浸渍搅拌并超声分散后进行冷冻干燥,得到了金属离子均匀分布的氧化石墨烯三维泡沫,经过在氨气/氩气气氛下的热分解,制备出单金属原子分散的三维氮掺杂石墨烯基催化剂材料(M1/NG,M=Fe,Co或Ni).将该M1/NG涂覆在商用PP隔膜上得到的改性隔膜显著提升了电池的电化学性能.研究发现,氮掺杂石墨烯上的金属单原子不但增强了材料对多硫化物的结合,从而有效阻止了多硫化物的穿梭,而且提升了多硫化物转化的氧化还原动力学,加速了多硫化物的转化.研究人员通过理论计算和电化学测试证实,与其它两种单原子相比,Fe SACs 对锂硫电池的电化学性能有更大的提升[图9(B)~(D)].Li2S6的可视化吸附实验展示了Fe1/NG对多硫化物更强的吸附作用.DFT计算显示,Li2S6分子在Fe1/NG中的Fe金属上的吸附比在NG中的氮掺杂剂上的吸附强(2.36vs.1.11 eV),且Fe-S(0.2248 nm)和N-Li(0.2135 nm)的距离明显缩短[图9(A)].隔膜渗透实验也进一步证实了Fe SACs对多硫化物的强吸附作用.为了探究SACs对多硫化锂催化转化的影响,在充放电过程中采用电化学原位拉曼光谱进行研究[图9(E),(F)].在2.8 V放电初始时,位于150.0,219.2 和475.0 cm-1处的拉曼信号分别对应S8物种的不对称弯曲、对称弯曲和对称拉伸.随着放电的进行,在2.42 V 时出现了(202.1 和445.3 cm-1)的特征峰,对应S8转化为长链多硫化物.在2.24 V时S8信号完全消失,代表其完全转化,而对照组的商业PP隔膜仍存在S8的信号,验证了Fe SACs 对多硫化锂转化的催化效果.在后续放电过程中,当电压从2.10 V 降低到1.80 V 时,Fe1/NG 改性PP 隔膜在约375.0 cm-1处的Li2S 峰占主导地位.而PP隔膜电池仍可以观察到的残留信号,这是由于直到放电结束后阴极中的硫仍然还原不完全造成的.在随后的充电过程中,当电压从1.72 V增加到2.60 V时,和S8的拉曼信号重新出现.
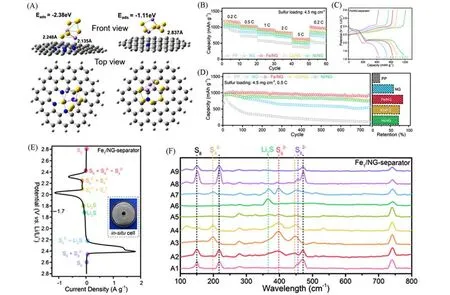
Fig.9 Theoretical calculation and experimental characterization of NG and Fe1/NG in Li⁃S batteries[57]
Xiong等[58]通过自模板及自还原策略制备了一种负载在氮掺杂石墨烯上且具有N和O双配位的W单原子催化材料[W/NG,图10(A)],并将其用于锂硫电池的隔膜改性,显著提升了电池的电化学性能.W单原子均匀锚定在石墨烯上,提供了大量的活性位点,而且通过调控金属源的加入量实现了W单原子高达8.6%的高负载量.该新型SACs具有独特的W-N2O2-C配位构型,W原子局域的特殊配位环境使得W/NG能更有效地锚定多硫化物,并对多硫化物的转化表现出更高的催化活性,即在增强对多硫化物吸附的同时,促进了长链LiPSs与产物Li2S之间的相互转化,有效地抑制穿梭效应.通过DFT理论计算[图10(B)~(D)]进一步证实,与常规的W-N4-C构型相比,具有强电负性的氧原子的引入一方面增加了体系的极性,使得W—S键强度提升,进而提高了对LiPSs的锚定效果;另一方面增加了体系与多硫化物之间的有效电荷转移,从而提升对LiPSs转化的催化能力.电池具有高的倍率性能,在10C倍率下的放电容量为678 mA·h·g-1,并可在2C 倍率下稳定循环1000 次.而且,在8.3 mg/cm2高硫负载下,实现了6.24 mA·h·cm-2的高面容量并保持了较好的循环稳定性[图10(E)和(F)].
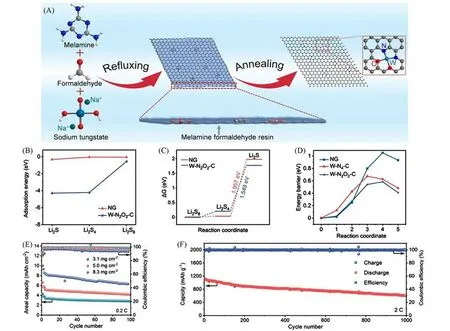
Fig.10 Schematic illustration and electrochemical performance of W/NG[58]
调控单原子碳载体结构能够有效改善单原子与多硫化物的相互接触,从而提高SACs 的利用率.Huang等[37]采用超分子curbituril骨架作为前体,通过热解得到了锚定在介孔碳的骨架内CoSACs材料.得益于介孔碳骨架丰富的孔隙结构和大的比表面积,锚定在其上的Co单原子能够与多硫化物充分接触并相互作用,大大提高了Co单原子作为吸附和催化活性位点的利用率.
通过巧妙的结构设计将SACs与其它材料杂化复合制备多功能中间层也是提高单原子催化材料利用率的有效策略.Guo等[59]通过逐层组装锚定了Co单原子的氮掺杂碳纳米片(NC@SA-Co)与高导电碳纳米管-纤维素纳米纤维双杂化网络制备了锂硫电池的自支撑多功能中间层图[图11(A)].由图11(B)可见,多功能中间层中的导电碳纳米管网络作为物理屏障限制多硫化物的扩散,进而促进了多硫化物的转化,同时富含端基氧的纤维素纳米纤维网络允许Li+离子快速输运,并通过强静电排斥力抑制多硫化物的穿梭.而且,NC@SA-Co 中大量的Co-N4位点作为高催化活性中心有效促进了多硫化物可逆转化的反应动力学,显著提升了电池的稳定性和循环寿命.因此,组装了多功能中间层改性隔膜的电池具有优异的电化学性能,在2C 倍率下稳定循环700 次,每次循环的容量衰减率低至0.058%.在7.2 mg/cm2的高硫负载情况下,锂硫电池能够实现8.3 mA·h·cm-2的高面容量.
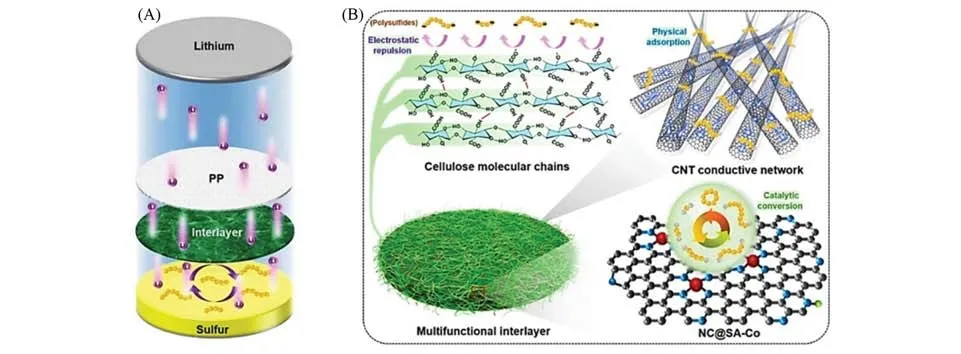
Fig.11 Schematic illustration of the multifunctional interlayer[59]
进一步提升SACs的单原子载量来增强其对多硫化物的吸附能力和催化活性,从而有效抑制多硫化物的穿梭和锂枝晶问题一直受到研究人员的广泛关注.针对这一问题,Guo等[60]设计了一种基于二维超薄钴金属有机框架纳米片(MOF-Co,约1.2 nm)构建的单原子阵列催化剂,并将其与细菌纤维素通过逐层组装的方式制备得到了双功能隔膜(B/2D MOF-Co),用于替代传统的PP隔膜作为锂硫电池的自支撑隔膜.在所得的自支撑功能化隔膜中,规则排列的Co 原子与O 原子发生配位(Co-O4)后暴露在MOF纳米片的表面而展现了单原子阵列状态.经能量色散X射线(EDX)光谱仪元素分析显示,钴原子的质量分数高达24.8%,接近基于晶体结构(Co-O4)的计算值(28.9%,质量分数).制备的B/2D MOF-Co隔膜轻质且具有柔韧性,对电解质有很好的浸润性,而且具有良好的热稳定性和机械性能.通过实验和理论计算分析发现:在负极侧,表面具有周期性排列的Co-O4结构和增强的机械模量的2D MOF-Co 纳米片可以通过强锂离子吸附使锂离子通量均匀化,有助于Li 的均匀成核并抑制Li 枝晶生长;在正极侧,2D MOF-Co纳米片可以通过路易斯酸碱相互作用有效锚定多硫化物,限制多硫化物扩散,提高硫利用率(图12).基于这种双功能单原子阵列(B/2D MOF-Co)隔膜组装的电池显示了良好的放电容量和循环稳定性.在高达7.8 mg/cm2的高硫载量下,组装的电池在循环200圈后的容量仍然高达5.0 mA·h·cm-2.研究人员也将该双功能隔膜用于柔性软包锂硫电池,组装的电池在多种弯曲角度下都显示出稳定的循环性能,说明了该双功能单原子阵列材料作为隔膜的应用潜力.
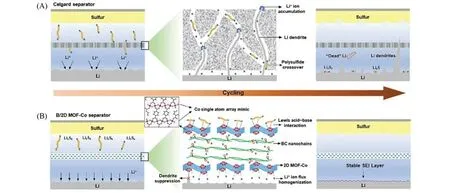
Fig.12 Schematic illustration for the Li⁃S batteries with celgard and B/2D MOF⁃Co separators[60]
由于单金属中心具有较高的表面自由能,单原子在化学上不稳定,倾向于聚集成金属纳米粒子,而且基于空间限制策略制备SACs的合成过程通常涉及到基体材料的复杂缺陷工程和高温热解,因此,开发有效的方法来获得稳定且具有较高催化性能的SACs也受到诸多研究者的关注.Lin等[61]通过原子层沉积(ALD)的方法在3 μm厚的碳纳米管薄膜上修饰了单原子分散的钴,制备得到了负载Co单原子的碳纳米管薄膜催化剂[CNT@SACo,图13(A)],并将其作为多功能中间层用于锂硫电池.通过实验和理论计算分析发现,CNT@SACo 催化剂中碳纳米管表面随机分布的Co 单原子对多硫化物具有强吸附能力,而且显著提升了多硫化物的电化学转化动力学[图13(B),(C)].修饰了CNT@SACo中间层的锂硫电池表现出高容量、长期循环稳定性和出色的倍率性能.由图13(D)可见,在1C的倍率下,电池能够提供高达880 mA·h·g−1容量,且能够稳定循环500次,每次循环的容量衰减率仅为0.064%.
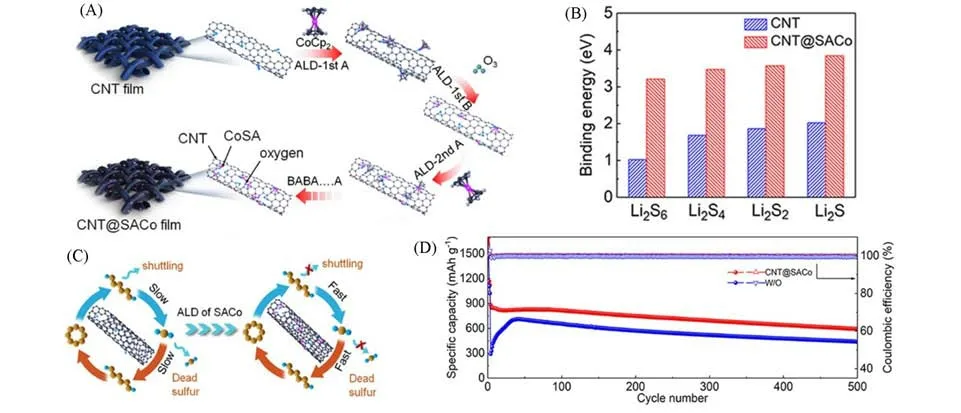
Fig.13 Schematic illustration and electrochemical performance of CNT@SACo interlayer[61]
4 总结与展望
本文综合评述了近年来SACs材料在锂硫电池的正极、负极、隔膜/中间层中的应用进展.SACs具有独特的电子结构和最大化的原子利用率,能增强对LiPSs的吸附,促进LiPSs的转化动力学,提高电池的倍率性能.而且SACs能作为亲锂位点可抑制锂枝晶的生长.虽然SACs在锂硫电池领域的研究取得了一些进展,但是仍有许多问题亟待解决以进一步推动SACs在锂硫电池中的广泛应用.(1)锂硫电池中的SACs的金属中心研究主要集中在Fe,Co以及Zn等元素,元素种类有限,其它金属元素和非金属元素被关注得较少或尚未被研究.(2)精确调节金属中心的配位环境,进一步提高活性中心的作用,除N,O和S外可考虑掺杂元素(如B和P等),以及增加空位作为SACs的锚定位置.(3)SACs作为锂硫电池的正极主体材料,载体除了影响SACs 的负载量和结构外,还应具备高导电性以快速传递电子、缓解体积膨胀、多孔互连结构增大硫的负载量以及物理限制LiPSs的穿梭等作用,因此还需要进一步优化载体材料的结构.(4)虽然目前对锂硫电池中的SACs的催化机理有了一定的认识,但更深入的反应机制目前仍不清楚,需要借助进一步的理论研究和原位表征方法来深入理解相关机制,为锂硫电池中的SACs设计提供理论指导.(5)关注在贫电解液、高载硫量等实际条件下SACs的应用,发展低成本、高负载的SACs的制备方法,推动单原子材料在锂硫电池中的实际应用.
