非洛地平纳米晶的制备及质量评价
2022-06-08李芳平
李芳平,邓 梦,汪 洋
武汉市红十字会医院药剂科,武汉 430015
非洛地平为二氢吡啶类钙拮抗剂,临床上主要用于高血压、缺血性心脏病、心力衰竭等病症的预防和治疗[1]。非洛地平在生物药剂学分类系统(BCS)中属于Ⅱ类药物,其在水中溶解度极低(<5.0 μg·mL-1)[2],口服吸收较差,生物利用度约为10%~20%[3]。纳米晶是通过特殊工艺将药物颗粒粒径减小至亚微米级来提高药物的溶出率[4],进而提高药物的生物利用度[5]。本研究采用介质研磨法制备非洛地平纳米晶,并根据单因素实验结果确定了纳米晶的制备工艺参数及处方组成,为非洛地平的二次开发利用提供新的研究思路。
1 仪器与试药
1.1 仪器
DRS2000高速高剪切乳化分散机(德国IKA公司);DYNO-Mill Research Lab介质研磨机(华尔宝机械有限公司);氧化锆珠粒(直径为0.4、0.6、1.0 mm);Malvern Zetasizer Nano ZSE纳米粒度电位仪(英国马尔文公司);FEI Nova 400型场发射扫描电子显微镜(美国FEI公司);RT600溶出度仪(深圳市锐拓仪器设备有限公司)。
1.2 试药
非洛地平(江苏联环药业股份有限公司,批号20200311,质量分数为99.5%);羟丙基纤维素SL(HPC SL),羟丙基纤维素SSL(HPC SSL),均购自日本曹达株式会社;羟丙甲纤维素E5(HPMC E5),羟丙甲纤维素E15(HPMC E15),均购自陶氏化学公司;聚乙烯醇(PVA,默克化工技术有限公司);聚乙烯己内酰胺聚醋酸乙烯酯聚乙二醇接枝共聚物(Soluplus®),聚乙烯吡咯烷酮K30(PVP K30),泊洛沙姆188(Poloxamer 188),十二烷基硫酸钠(SDS),均购自德国巴斯夫公司;吐温-80(Tween-80,湖南新绿方药业有限公司);维生素E聚乙二醇琥珀酸酯(TPGS,上海昌为医药辅料技术有限公司);去离子水(实验室自制);非洛地平片(规格:5 mg,阿斯利康制药公司)。
2 方法与结果
2.1 非洛地平纳米晶的制备
介质研磨法是制备药物纳米晶最成熟的技术之一,目前已有多个药品采用该技术进行了商业化生产[6],因此本研究采用介质研磨法制备非洛地平纳米晶。使用气流粉碎机将非洛地平原料药粉碎至粒径(D90)约为5~10 μm,称取一定量原料药加入到100 mL含有稳定剂的介质溶液中,分散均匀后经高剪切乳化机(剪切速度为10 000 r·min-1)剪切分散10 min,之后转移到介质研磨机中,加入一定量的氧化锆珠粒以一定的速度进行研磨,即得到非洛地平纳米晶。
2.2 制备工艺及处方筛选
2.2.1研磨介质体积考察 以高分子聚合物HPC SL作为非洛地平纳米晶的稳定剂,质量浓度为20 mg·mL-1,药物质量浓度为100 mg·mL-1,选择直径为0.4 mm的氧化锆珠粒作为研磨介质,研磨速度为2 000 r·min-1,研磨时间为2 h,分别考察药物溶液体积与研磨介质体积比为1∶0.2、1∶0.4、1∶0.6、1∶0.8、1∶1.0、1∶1.2对非洛地平纳米晶的粒径分布、多聚分散系数(PDI)和Zeta电位的影响。结果见表1。

表1 研磨介质体积考察实验结果
实验结果显示,增加研磨介质用量有助于减小非洛地平纳米晶的粒径,当药物溶液体积与研磨介质体积比为1∶1时,制备的非洛地平纳米晶粒径最小,这是由于研磨介质用量较高时,药物颗粒可以充分地分散到氧化锆珠粒之间的间隙中,能够产生有效的研磨作用[7],因此固定药物溶液体积与研磨介质体积比为1∶1.0。
2.2.2研磨介质直径考察 以高分子聚合物HPC SL作为稳定剂,质量浓度为20 mg·mL-1,药物质量浓度为100 mg·mL-1,药物溶液体积与研磨介质体积比为1∶1.0,研磨时间为2 h,研磨速度为2 000 r·min-1,考察氧化锆珠粒直径分别为0.4、0.6、1.0 mm对非洛地平纳米晶的粒径分布、PDI和Zeta电位的影响。结果见表2。
表2 研磨介质直径考察实验结果
实验结果显示,使用直径为0.4 mm的氧化锆珠粒作为研磨介质制备的非洛地平纳米晶粒径最小,这是由于较小直径的氧化锆珠粒有助于与药物颗粒充分接触,从而发挥较好的研磨作用[8],由于目前市场上只能购买到最小直径为0.4 mm的氧化锆珠粒,因此采用直径为0.4 mm的氧化锆珠粒进行实验。
2.2.3研磨时间考察 以高分子聚合物HPC SL作为稳定剂,质量浓度为20 mg·mL-1,药物质量浓度为100 mg·mL-1,药物溶液体积与研磨介质体积比为1∶1.0,研磨介质氧化锆珠粒直径为0.4 mm,研磨速度为2 000 r·min-1,考察研磨时间分别为1、2、4、6 h对非洛地平纳米晶的粒径分布、PDI和Zeta电位的影响。结果见表3。
表3 研磨时间考察实验结果
实验结果显示,增加研磨时间有助于减小非洛地平纳米晶的粒径,但是研磨时间超过4 h后纳米晶的粒径出现增加趋势,这是由于粒径足够小的纳米晶具有极高的表面自由能,研磨时间继续延长会出现奥斯特瓦尔德熟化现象,导致粒度增加[9],因此固定研磨时间为4 h。
2.2.4研磨速度考察 以高分子聚合物HPC SL作为稳定剂,质量浓度为20 mg·mL-1,药物质量浓度为100 mg·mL-1,药物溶液体积与研磨介质体积比为1∶1.0,研磨介质氧化锆珠粒直径为0.4 mm,研磨时间为4 h,考察研磨速度分别为1 000、1 500、2 000、2 500 r·min-1对非洛地平纳米晶的粒径分布、PDI和Zeta电位的影响。结果见表4。
表4 研磨速度考察实验结果
实验结果显示,增加研磨速度有助于减小非洛地平纳米晶的粒径,在所研究的研磨速度范围内,速度达到2 500 r·min-1时所制备的纳米晶粒径最小,而继续增加研磨速度会导致氧化锆珠粒之间发生剧烈碰撞,珠粒碎裂,污染药物[10],因此固定研磨速度为2 500 r·min-1。
2.2.5药物质量浓度考察 以高分子聚合物HPC SL作为稳定剂,质量浓度为20 mg·mL-1,药物溶液体积与研磨介质体积比为1∶1.0,研磨介质氧化锆珠粒直径为0.4 mm,研磨时间为4 h,研磨速度为2 500 r·min-1,考察药物质量浓度分别为50、75、100、125、150 mg·mL-1对非洛地平纳米晶的粒径分布、PDI和Zeta电位的影响。结果见表5。
表5 药物质量浓度考察实验结果
实验结果显示,随着药物质量浓度的增加,制备的非洛地平纳米晶的粒径呈减小趋势,这是由于药物质量浓度增加,提高了药物颗粒与氧化锆珠粒接触概率,药物质量浓度达到125 mg·mL-1时制备的非洛地平纳米晶的粒径最小,因此非洛地平纳米晶的处方中固定药物质量浓度为125 mg·mL-1。
2.2.6稳定剂种类考察 固定药物质量浓度为125 mg·mL-1,药物溶液体积与研磨介质体积比为1∶1.0,研磨介质氧化锆珠粒直径为0.4 mm,研磨时间为4 h,研磨速度为2 500 r·min-1,分别考察以HPC SL、HPC SSL、HPMC E5、HPMC E15、PVA和PVP K30作为稳定剂(质量浓度均为20 mg·mL-1)对非洛地平纳米晶的粒径分布、PDI和Zeta电位的影响。结果见表6。
表6 稳定剂种类考察实验结果
实验结果显示,与使用HPC SL、HPMC E5、HPMC E15、PVA和PVP K30作为稳定剂相比,采用HPC SSL作为稳定剂制备的非洛地平纳米晶的粒径更小,一方面是由于HPC SSL可以降低纳米晶表面与介质溶液表面之间的界面张力,使药物纳米晶被充分润湿,另一方面HPC SSL的黏度较低,黏性阻尼效应也较低[11],在研磨时能够有效地吸附在药物粒子表面,阻止其聚集、吸附,因此选用HPC SSL作为稳定剂进行实验。
2.2.7稳定剂质量浓度考察 固定药物质量浓度为125 mg·mL-1,药物溶液体积与研磨介质体积比为1∶1.0,研磨介质氧化锆珠粒直径为0.4 mm,研磨时间为4 h,研磨速度为2 500 r·min-1,选择HPC SSL作为稳定剂,考察其不同质量浓度对非洛地平纳米晶的粒径分布、PDI和Zeta电位的影响。结果见表7。
表7 稳定剂质量浓度考察实验结果
实验结果显示,随着HPC SSL质量浓度的增加,制备的非洛地平纳米晶的粒径呈减小趋势,但是其质量浓度超过20 mg·mL-1后纳米晶的粒径呈增加趋势,这是由于HPC SSL在质量浓度较低时,没有足够量的稳定剂吸附到纳米晶表面,纳米晶容易吸附聚集,当HPC SSL的质量浓度足够高后,体系的黏度增加,研磨时黏性阻尼效应较强,反而不利于粒径的减小[11],因此非洛地平纳米晶的处方中固定HPC SSL质量浓度为10 mg·mL-1。
2.2.8表面活性剂种类考察 虽然处方中的HPC SSL可以在一定程度上降低固液界面张力,增加润湿性[12],但是其润湿能力有限,需要在处方中加入表面活性剂来进一步降低界面张力[13]。因此本研究对表面活性剂种类进行筛选。以高分子聚合物HPC SSL作为稳定剂,质量浓度为10 mg·mL-1,同时分别在介质溶液中加入表面活性剂SDS、Poloxamer 188、Tween-80、Soluplus和TPGS(质量浓度均为0.75 mg·mL-1)作为稳定剂,固定药物质量浓度为125 mg·mL-1,药物溶液体积与研磨介质体积比为1∶1.0,研磨介质氧化锆珠粒直径为0.4 mm,研磨时间为4 h,研磨速度为2 500 r·min-1,考察处方中加入不同种类表面活性剂对非洛地平纳米晶的粒径分布、PDI和Zeta电位的影响。结果见表8。
表8 不同种类表面活性剂考察实验结果
实验结果显示,在处方中加入TPGS可制备出粒径更小且粒径分布更均匀的非洛地平纳米晶,因此加入TPGS作为表面活性剂进行实验。
2.2.9表面活性剂质量浓度考察 以高分子聚合物HPC SSL作为稳定剂,质量浓度为10 mg·mL-1,固定药物质量浓度为125 mg·mL-1,药物溶液体积与研磨介质体积比为1∶1.0,研磨介质氧化锆珠粒直径为0.4 mm,研磨时间为4 h,研磨速度为2 500 r·min-1,考察处方中加入不同质量浓度表面活性剂TPGS对非洛地平纳米晶的粒径分布、PDI和Zeta电位的影响。结果见表9。
表9 表面活性剂质量浓度考察实验结果
实验结果显示,随着处方中TPGS质量浓度的增加,制备的非洛地平纳米晶粒径呈先减小后增大趋势,这是由于TPGS的质量浓度较低时,药物粒子表面未完全被润湿,因此制备的纳米晶粒径较大,但是当表面活性剂质量浓度增加到一定程度后会形成胶束[14],对药物起到增溶作用,因此非洛地平纳米晶的处方中TPGS质量浓度固定为1.00 mg·mL-1。
2.3 纳米晶质量评价
2.3.1粒径分布和Zeta电位测定 根据2.2项下非洛地平纳米晶的制备工艺与处方单因素考察实验结果制备3批样品,取少量样品加入去离子水适当稀释,通过Malvern Zetasizer Nano ZSE纳米粒度电位仪检测其粒径分布和PDI(测量的环境温度为25 ℃,散射角为90°);另取少量样品加入去离子水适当稀释,通过电泳迁移率法测定Zeta电位,测定结果见图1。实验结果显示,3批非洛地平纳米晶的粒径平均值为(226.3±10.9) nm,PDI为(0.188±0.005),Zeta电位为(-25.1±0.5) mV。

图1 非洛地平纳米晶的粒径分布和Zeta电位
2.3.2扫描电镜观察 取新制备的非洛地平纳米晶溶液,加入去离子水适当稀释,摇匀,取一小滴样品均匀地铺展到锡箔纸表面,自然风干,通过导电胶带将样品黏附到样品台上,喷金,在扫描电镜下观察非洛地平纳米晶的微观形态,并拍摄照片,见图2。在扫描电镜下可观察到非洛地平纳米晶呈不规则状,粒径在100~400 nm范围内分布。

图2 非洛地平纳米晶扫描电镜图
2.3.3体外溶出度研究 通过体外溶出实验比较非洛地平纳米晶和非洛地平片的体外药物溶出情况,选择桨法,转速为50 r·min-1,溶出介质为pH1.2盐酸溶液(Tween-80质量浓度为0.2 mg·mL-1),体积为500 mL,温度为37 ℃±0.5 ℃,分别取非洛地平纳米晶和非洛地平片加入到溶出杯中,并在预设的时间间隔取出溶出介质5 mL,并补加相同体积的空白溶出介质(37 ℃),将取出的溶液经0.1 μm 微孔滤膜过滤,通过HPLC法进行定量分析,绘制药物溶出度时间曲线。见图3。
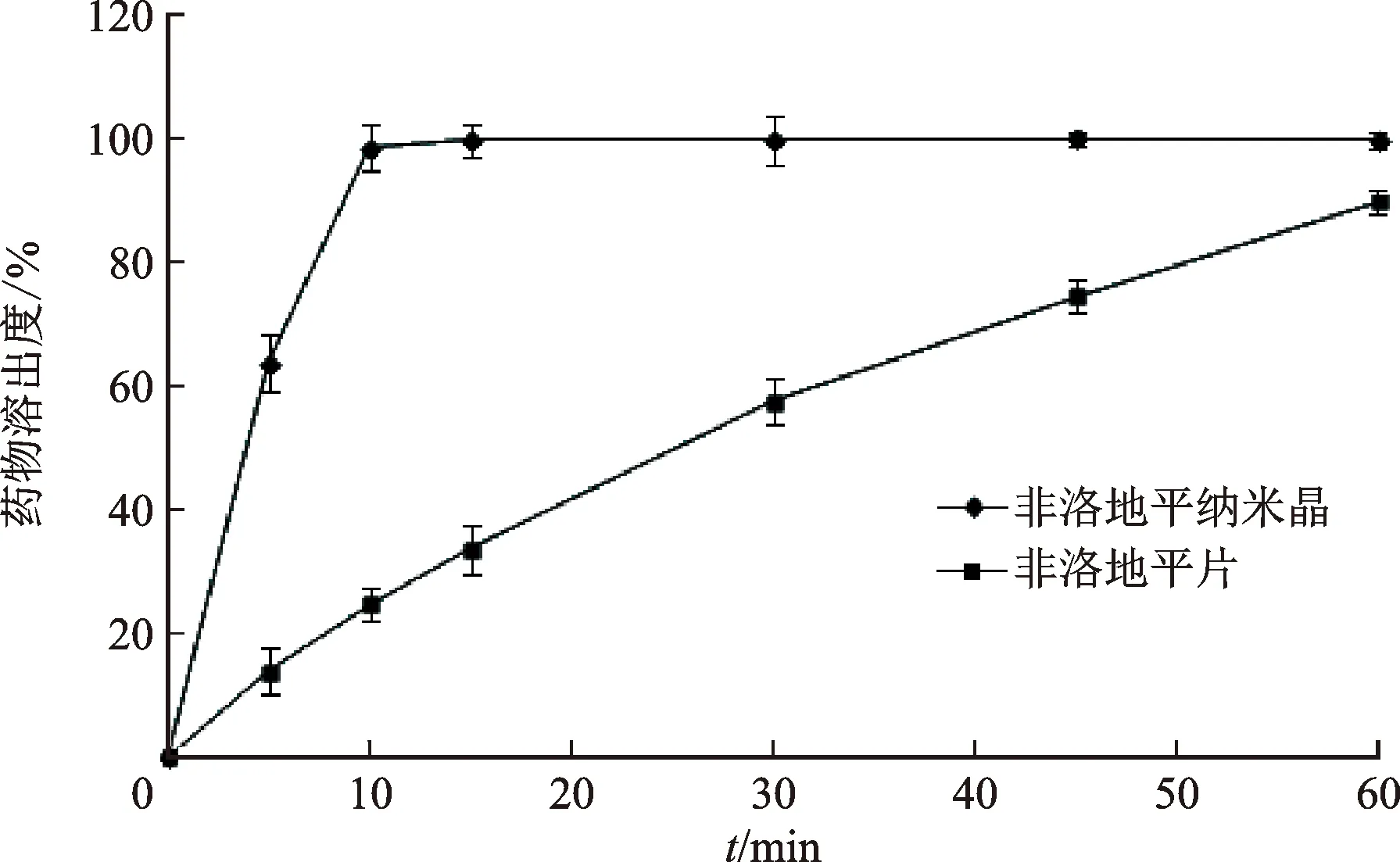
图3 非洛地平纳米晶和非洛地平片的体外溶出度时间曲线
体外溶出实验结果显示,非洛地平纳米晶中药物在5 min溶出度为63.6%,10 min内可达到100%溶出,而相同时间内非洛地平片中的药物溶出度仅有13.9%、24.7%,直到60 min时药物溶出度才达到89.3%,与非洛地平片相比,非洛地平纳米晶中的药物溶出速度和程度均得到显著提高。
3 讨论
由于纳米晶药物粒子具有极高的比表面自由能,属于热力学的不稳定体系,药物粒子存在聚集倾向[15],纳米晶的处方中需要加入稳定剂以阻止其相互聚集[16]。本文制备的非洛地平纳米晶,经过筛选确定在处方中加入HPC SSL和TPGS作为稳定剂,HPC SSL可吸附在纳米晶表面,通过空间位阻作用阻碍纳米晶相互聚集,而TPGS不仅可以降低液固界面张力,润湿药物粒子表面,而且可通过氢键作用吸附到纳米晶体表面,提供机械屏障[17],进一步抑制纳米晶体聚集。
本文通过全面、系统地对非洛地平纳米晶的制备工艺及处方进行研究,虽然其溶出度得到了显著的提高,但是非洛地平纳米晶为液体状态,其储存及患者服用均存在一定的问题,纳米晶最终需要通过固化工艺制备成稳定的、便于口服的固体制剂[18],因此本课题组后期对非洛地平纳米晶的固体化进行相关研究。
