抗结核药物靶点的研究进展
2022-05-30吴疆崔嘉伟薛云新王岱
吴疆 崔嘉伟 薛云新 王岱


摘要:由结核分枝杆菌(Mycobacterium tuberculosis,Mtb)感染引起的结核病(TB)是世界上最致命的感染性疾病之一。因抗结核药物的泛用与滥用,细菌耐药问题日渐凸显。近几十年仅有2种抗结核新药上市,由于药物使用中的副作用及其耐药菌株的出现,急需开展针对Mtb新靶点的药物研究。本文围绕近几年全球最新报道的抗结核药物靶点及其相关化合物进行综述并加以系统总结,以期在一定程度上为新型抗结核药物的研发提供参考。
关键词:结核分枝杆菌;抗结核药;靶点
中图分类号:R978.3 文献标志码:A
Advances in targets of antituberculosis drugs
Wu Jiang, Cui Jia-wei, Xue Yun-xin, and Wang Dai
(State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics, Xiamen University, Xiamen 361102)
Abstract Tuberculosis (TB) is one of the world's most deadly infectious diseases caused by Mycobacterium tuberculosis (Mtb). Due to the widespread use and abuse of anti-tuberculosis drugs, the problem of bacterial drug resistance is becoming increasingly prominent. In recent decades, there are only two new anti-tuberculosis drugs in the market, due to the side effects of drug use and the emergence of drug-resistant strains, there is an urgent need to develop new drug targets for Mtb. In this paper, the targets and related compounds of anti-tuberculosis drugs reported in recent years are reviewed and systematically summarized in order to provide a reference for the research and development of new anti-tuberculosis drugs to some extent.
Key words Mycobacterium tuberculosis; Antituberculosis drugs; The target
由結核分枝杆菌(Mycobacterium tuberculosis, Mtb)导致的结核病(tuberculosis, TB)是现今对人类最具致命性的一类传染性疾病。世界卫生组织(WHO)的最新统计数据显示[1],2020年有990万TB病患并有约150万例病患因此死亡。尽管临床上已有可以预防和治疗TB的药物,但是由于近年来抗结核药物的泛用和滥用,导致多重耐药甚至广泛耐药结核分枝杆菌的大量出现,使得临床治疗面临难以解决的困境[2]。
1 抗结核药物现况
自20世纪40年代起,链霉素(streptomycin,SM) 和对氨基水杨酸(p-amino salicylic acid,PAS) 先后被发现并用于治疗TB。随着1952年异烟肼(INH)的出现,形成“INH+SM+PAS”的三联疗法,一般需要18~24个月的治疗期。基于患者的药源和耐受性,其中的PAS可选用氨硫脲(TB1)或乙胺丁醇(EMB)替换。1965年利福平(RIF)开始应用于临床后,相关研究发现“INH+RIF”可将该疗程减短到9个月,再进一步与吡嗪酰胺(PZA)结合使用能够使疗程减短至半年[3]。此后,相关研究一直没有研发出应用于TB治疗的全新结构药物。
按照其杀菌活性、安全性、临床疗效与药品价格,可将抗结核药物划分成一线和二线药物。一线药物相对疗效好,副作用小且价格较低,目前对药物敏感性TB患者的治疗首选仍是INH、EMB、RIF等一线药物[4](表1)。当病原体对一线药物产生耐受时,治疗可用氟喹诺酮类药物,如利奈唑胺(LZD)、莫西沙星(MXF)、左氧氟沙星(LVFX)等(表2)。面对广泛耐药和已有二线药物耐药结核分枝杆菌的威胁,在过去10年中人们研发推出了两种二线新药:贝达喹啉和德拉马尼,其相关的临床试验和应用使得TB的治疗有所改善[5]。但由于其诸多使用条件和易产生不良反应的限制,并不能从根本上解决抗结核药物匮乏的问题。该现状促使研究人员深入了解Mtb的生理特性及其致病机制,以期发现新的抗结核靶点和进一步研发新型抗结核药物。
2 抗结核药物靶点
2.1 细胞壁合成相关
Mtb的细胞壁与其致病性和侵袭力相关,其中许多成分可能成为新型抗结核药物的关键靶点。
2.1.1 DprE(decaprenylphosphoryl-D-ribose 2'-epimerase)
DprE (decaprenylphosphoryl-D-ribose 2'-epimerase) 是由DprE1和DprE2构成的异二聚体,在D-核糖向D-阿拉伯呋喃糖(decaprenyl-phosphoryl D-arabinose,DPA) 的转化中起重要作用,而DPA是合成细胞壁阿拉伯聚糖的唯一前体。目前研发的抑制剂主要针对DprE1,包括苯并噻嗪酮类、氟喹诺酮类和氮杂吲哚类,其中氮杂吲哚衍生物TBA-7371处于临床试验的第一阶段。Suma等[6]使用基于结构的药效团模型对氮杂吲哚衍生物进行筛选,并通过诱导匹配对接和分子动力学结合Prime MM-GBSA计算进一步验证,从ZINC数据库中筛选出两种化合物后将结果与临床试验的抑制剂比较,得到了一个对DprE1具有最大抑制潜力的化合物ZINC000170252277(图1)。
2.1.2 分枝杆菌膜蛋白3
作为跨膜转运蛋白,分枝杆菌膜蛋白3 (mycobacterial membrane protein large 3,Mmp L3) 主要参与Mtb的脂质运输、聚合和机体免疫调节等功能,是近期发现的一类治疗药物靶点[7]。通过实验已筛选出一系列作用于该靶点的化合物(如SQ109、AU1235、BM212等),其中二胺类衍生物SQ109(图2)的最小抑菌浓度(minimum inhibitory concentration,MIC)范围为0.12至0.78 mg/L,耐受性良好且在动物模型中可以达到药代动力学/药效学目标[8],已进入临床试验的第二阶段。
2.1.3 L-鼠李糖合成相关酶
L-鼠李糖是一种脱氧己糖,主要功能是将细胞壁中两种成分肽聚糖和阿拉伯半乳糖连接起来,导致Mtb无法正常合成细胞壁。由于人类体内没有L-鼠李糖成分,因此其合成相关酶成为可供选择的药物靶点[9]。L-鼠李糖在Mtb内的合成来源主要通过4种酶促反应,参与反应的酶分别是Rml A (glucose-L-phosphate thymidylyltransferase)、Rml B (dTDP-glucose-4,6-dehydratase)、Rml C (dTDP-6-deoxy-D-xylo-4-hexulose-3,5-epimerase)和Rml D (dTDP-6-deoxy-L-lyxo-4-hexulose C4-reductase)。其中Rml C因其结构的特殊和底物的高特异性,以及对辅酶因子无依赖性而在研究领域受到广泛关注,但目前尚无相关的抑制剂上市。Rath等[10]提出大黄酚可作为Rml C抑制剂的候选化合物。而Dhaked等[11]通过对4个酶进行蛋白质结构建模并分析,筛选出G8、Q80、G85等几十个具有潜在功能的残基以探讨催化机制,以期对未来相关化合物的合成提供参考方向。
2.1.4 烯酰基载体蛋白还原酶
烯酰基载体蛋白还原酶(Inh A)在细胞壁脂肪酸合成方面发挥着重要作用,是一线药物INH的一个关键作用靶点[12]。INH属于一类前体药物,依赖于KatG酶将其激活,而KatG的基因突变会阻断该激活途径并导致耐药。因此现在许多研究聚焦于Inh A的“直接抑制剂”。三氯生(TCS)属于一类广谱抗生素,可对Inh A造成可逆、温和的抑制。Armstrong等[13]应用计算机设计、合成和表征了一系列带有连接到B环的1,5-三唑基团的新型TCS衍生物,从中筛选得到效果最佳的化合物11(图3),其MIC为(12.9±5.0) ?mol/L。然而其体外酶活显示效果并不理想,50 ?mol/L该化合物仅能达到11%的抑制效率。
2.1.5 D-丙氨酸-D-丙氨酸连接酶A
D-丙氨酸-D-丙氨酸连接酶A(D-Alanyl-D-alanine ligase A,DdlA)能在ATP的介导下促进D-丙氨酸 (Ala)分子的二聚化,获得D-丙氨酰-D-Ala。其产物与肽聚糖(PG)的五肽生物合成相关,能显著影响Mtb细胞壁的正常功能[14]。D-环丝氨酸(DCS)是DdlA的抑制剂,其添加(16 μg/mL)可以抑制菌株的生长。Yang等[15]建立了1种TB-DdlA活性比色法用于DdlA抑制剂的高通量筛选,同时采用pull-down实验和MS/MS分析了TB-DdlA之间的相互作用,并据此鉴定出8个不同的TB-DdlA潜在的相互作用蛋白。Meng等[16]通过高通量筛选出了DdlA竞争性抑制剂IMB-0283(图4),其对标准和临床耐药Mtb菌株的MIC范围为0.25~4.00 μg/mL,且在小鼠模型中可以检测到良好的抑菌活性。但是抑制酶活和抑菌活性的实验结果存在差异,表明DdlA可能不是IMB-0283的主要靶点,这仍需进一步研究其作用机制以保证其体内应用的安全性。
2.1.6 HspX蛋白
HspX蛋白是一种仅在Mtb中表达的α-晶体蛋白。研究发现,在宿主巨噬细胞中,低氧水平会诱导分枝杆菌细胞壁中的HspX高度表达。这表明它在TB感染的潜伏期中起着重要作用[17]。由于治疗期间人体容易产生抗生素耐药性,因此通过接种疫苗进行早期预防是控制感染的有效途径之一。目前可用的结核病疫苗只有卡介苗(Bacillus calmette-guerin,BCG)。Moradi等[18]通过编码融合HspX-PPE44-EsxV抗原,得到了一种新型DNA疫苗,并在BALB/c小鼠感染模型中分别测试了单独使用新型疫苗及其与BCG聯用后的效果。实验结果表明,HspX融合疫苗可以在小鼠体内诱导高水平的特定细胞因子,而且在BCG增强免疫方案中使用该DNA疫苗可产生大量的IFN-γ、IL-12和TGF-β等细胞因子,从而取得更好的疗效。
2.2 DNA/RNA合成相关
2.2.1 DNA促旋酶
DNA促旋酶是一种四聚体蛋白,由2个GyrA和2个GyrB亚基构成,以ATP依赖性方式催化DNA的负超螺旋[19]。由于其不存在于人体且是Mtb中唯一的II型拓扑异构酶,因此成为抗结核药物中的研究靶点。近年来,由于GyrA亚基易发生氟喹诺酮类耐药突变,药物研究及靶点筛选已转向GyrB亚基[20]。Rajput等[21]通过对用过的药物进行重新筛选,从中发现了松果菊苷、多柔比星、表柔比星和伊达比星4种对促旋酶B亚基具有高亲和力的药物(图5)。通过荧光光谱、圆二色谱法等测定,表柔比星和松果菊苷对Mtb的MIC90(试验中抑制90%细菌的MIC)最低,分别为6.3和12 μmol/L。同时,表柔比星和松果菊苷能够靶向DNA促旋酶,通过抑制其催化循环从而抑制分枝杆菌生长,这也为可用于耐药菌株的新型药物优化提供了理论基础。
2.2.2 RNA聚合酶
RNA聚合酶在Mtb和真核生物中的结构存在一定差别。抗生素RIF能与RpoB蛋白(RNA聚合酶的β-亚基)特异性结合,抑制RNA转录与合成,实现杀菌效果[22],因此编码RpoB蛋白的基因突变能导致Mtb对RIF耐药。RpoB蛋白基因中大部分突变位点处于81 bp的RIF抗性决定区,因而近年来针对利福平抗性决定区(rifampicin resistance determining region,RRDR) 的研究成为了热点方向。Hameed等[23]发现RpoB蛋白的531号氨基酸密码子突变同RIF的耐药存在联系。Zaw等[24]也发现RRDR中最常见的突变密码子为516号、526号和531号,其中531号位点氨基酸的突变发生最为广泛。目前需要能够识别与耐药突变相关的分子检测方法,以便进一步开展耐药突变的研究。
2.2.3 次黄嘌呤鸟嘌呤磷酸核糖转移酶
次黄嘌呤鸟嘌呤磷酸核糖转移酶(hypoxanthine-guanine phosphoribosyl transferase,HGPRT)是一种细胞质酶,它合成的6-氧嘌呤核苷单磷酸是DNA/RNA合成过程中所必需的[25]。两种吡咯烷核苷磷酸盐是目前针对Mtb-HGPRT最有效的抑制剂,Eng等[26]分别将Mtb和人体的HGPRT与从抑制剂中提取的一种复合物6作用后的晶体结构进行比较,该结果解释了抑制剂在体外对这两种酶抑制常数存在的60倍差异。复合物6的四酰胺前药(图6)能够抑制Mtb的生长,其在复制期半抑制浓度(half maximal inhibitory concentration,IC50)为14 μmol/L,而在潜伏期IC50为29 μmol/L。此外,它还能抑制Mtb在宿主巨噬细胞中的生长(MIC50为85 μmol/L),同时对哺乳动物细胞具有较低的毒性,其细胞半数毒性浓度(half cytotoxic concentration,CC50)为(132±20) μmol/L。此类化合物日后有望成为新型抗结核药物的前体化合物。
2.2.4 胸苷酸激酶
胸苷酸激酶(thymidine monophosphate kinase,TMK)是一种核苷单磷酸激酶,在嘧啶合成中起关键作用,对细菌的DNA合成至关重要[27]。TMK可催化ATP和脱氧胸苷-5'-单磷酸(dTMP)之间的可逆磷酸化,产生ADP和脱氧胸苷-5'-二磷酸(dTDP),并通过再次磷酸化得到胸苷5'-三磷酸(dTTP)[28]。Mtb-TMK在人体中的低同源性使其成为潜在的药物靶点。不仅Wayengera等[29]通过对TB患者样本的免疫血清检测发现Mtb-TMK可作为潜伏性感染的血清标记物。Naik等[30]也通过高通量筛选等方法,发现了氰基吡啶酮类和萘啶类化合物都对TMK具有较好的选择性。最新报道Venugopala等[31]通过计算机设计并评估了几种嘧啶酮和嘧啶硫酮化合物,最终嘧啶酮1a和嘧啶硫酮2a(图7)被筛选出作为针对TMK靶点的先导化合物。
2.3 能量代谢相关
2.3.1 ATP合成酶
Mtb中的ATP合酶是一种含有多个亚基的跨膜蛋白复合物,参与几乎所有活细胞的能量代谢和部分细胞间的信号传递[32]。由atpE基因编码的ATP合酶C亚基是抑制剂贝达喹啉的主要作用靶点。但基因突变导致的贝达喹啉耐受加上贝达喹啉所导致的心律失常及恶心、头痛一系列副作用,限制了其临床应用。Saxena等[33]开发了以计算参数为自变量,以活性 (-log IC50/MIC) 为因变量的定量构效关系 (quantitative structure-activity relationship,QSAR)模型,对一系列双喹啉衍生物进行了评估。Dhulap等[34]也对二芳基喹啉抑制剂的构效关系(structure-activity relationship,SAR)和临床药理学进行了评估,发现贝达喹啉SAR研究衍生的TBAJ-587和TBAJ-876化合物(图8)在体外和小鼠结核病模型研究中均显示出其对TB的潜在疗效,目前两类化合物皆处于临床研究的第一阶段。
2.3.2 细胞色素bc1: aa3
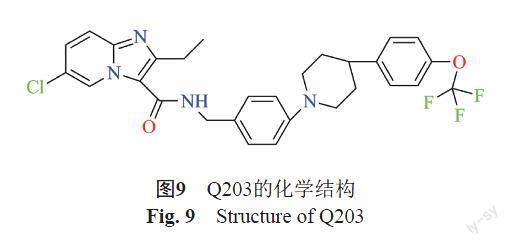
Mtb的细胞色素bc1: aa3是由甲萘醌、细胞色素c还原酶(bc1)和细胞色素aa3型氧化酶组成的复合体,主要在呼吸链中发挥电子传递效能,显著影响ATP的合成[35]。咪唑并[1, 2-α]吡啶类化合物Q203(图9)是针对细胞色素bc1: aa3复合体中细胞色素b亚基 (the cytochrome b subunit,QcrB)而研发的抑制剂,目前处于临床研究的第二阶段。研究发现Q203仅有抑菌功能而无法杀菌,这是因为Mtb的电子传递链内含细胞色素bc1: aa3、细胞色素bd氧化酶这2个末端氧化酶,Q203靶向QcrB将细胞色素bc1: aa3作用阻断的同时,细胞色素bd氧化酶能补偿ATP的合成从而保证细菌的存活。Kalia等[36]对细胞色素bd氧化酶的编码基因cydAB进行敲除后, 发现Q203能够完全抑制细菌的呼吸,這提示该类药物研发可以考虑从两种末端呼吸酶的相互作用入手。Cleghorn等[37]也鉴定发现靶向QcrB的新型吗啉-噻吩(morpholino thiophenes,MOT) 具有细胞低毒性、杀菌高活性[对H37Rv的MIC为(0.72±0.30) μmol/L],同时在急性感染小鼠模型中取得了成效。
2.3.3 莽草酸激酶
莽草酸途径的终产物是分支酸盐,这是一种芳香族氨基酸和甲基萘醌等物质的前体。在该途径中起关键作用的莽草酸激酶 (shikimate kinase,SK),近年来因其不存在于人体,但存在于包括Mtb在内的多种细菌内,成为药物靶点研究的对象[38]。Masoko等[39]从Sutherlandia frutescens(一种传统抗结核药物,具体作用机制未知)中分离出α-亚麻酸并在体外验证了其对SK的抑制效果(IC50为0.1 μg/mL)。Rahul等[40]应用了一种基于配体和结构的虚拟筛选方法,作为鉴定用于开发SK抑制剂的支架。并根据合成可行性选择了17种化合物测试其生物活性,结果显示这些化合物都有体外抑制酶活的作用。其中大多数化合物IC50为50 μg/mL,少数为25 μg/mL。尽管目前对SK的研究大多停留在计算机的模拟和筛选阶段,但合成的化合物往往与SK有较高的亲和力,也预示了SK作为抗结核药物靶点的广阔前景。
2.3.4 細胞色素P450酶系
细胞色素P450 (cytochromeP450,CYPs) 是一类具有混合功能的氧化还原酶,在细胞合成与分解代谢活动中发挥关键作用。CYPs在大多数细菌中不超过5种,甚至不存在于大肠杆菌中。但H37Rv基因组测序结果显示,在其基因组中有20种CYPs单氧酶[41]。氯法齐明(Cfz)被视作核心二线药物,体外鉴定结果显示其可有效抑制CYPs。Sangana等[42]发现在治疗TB感染过程中联用药物含有CYPs的底物(如米达唑仑)时,会发生相关代谢反应而对人体产生危害。正在进行Ⅱ期临床试验的SQ109是被看好的抗结核候选药物之一,已被验证能被人、狗和鼠的肝微粒体有效代谢。虽然目前还没有Mtb中CYPs与其相互作用的报道, 但Bukhdruker等[43]发现CYP124会在体外结合SQ109并羟基化后形成稳定的结构,因而作出SQ109只是前体,需要CYPs参与代谢后才能产生抗结核作用的假设,这也是首次发现Mtb中的CYPs能够将抗结核药物进行生物转化。
2.4 其他抗结核靶点
2.4.1 铁代谢相关
铁对Mtb的致病性和毒力是必不可少的。细菌从宿主蛋白内摄取铁时,铁载体发挥着非常重要的作用。分枝杆菌表达的腺苷化酶MbtA,对菌蛋白生物合成的初始反应具有催化作用。Ferguson等[44]测试了846种抑制Mtb生长的化合物与MbtA的结合能力并筛选得到效果最佳的一个化合物,发现其对MbtA具有高亲和性,MIC90值为13 μmol/L。Bythrow等[45]研发了一种水杨酸腺苷酸化酶抑制剂salicyl-AMS(图10)及其衍生物,可以靶向MbtA,并在小鼠模型中验证了其疗效(MIC为0.8至5.3 ?g/mL)。然而该药物的半衰期较短(50 mg/kg,腹腔给药时t1/2=13 min)及生物利用率低仍是限制其应用于治疗的短板。
铁依赖性调节蛋白(iron-dependent regulator,IdeR)在Mtb铁代谢当中有着重要作用。对IdeR的晶体学研究发现,仅有其二聚体结构具有完整的功能活性,能与DNA上保守区域相互作用。因此其抑制剂的研发方向主要是通过阻止二聚体的形成或阻断IdeR与DNA相关序列的相互作用。Salimizand等[46]尝试引入了两种相关新化合物(有效肽结构为RPR和GVPG)并通过体外抑菌实验验证其效力,该结果说明在抗结核药物研发方面,IdeR靶点具有相当的研究和应用潜力。
2.4.2 免疫相关
TB的临床治疗难点不仅在于耐药菌的出现,还有Mtb特有的免疫逃避机制。作为一类肽聚糖水解酶,RipA通过活化NFκB信号途径,同时促进IL-6、TNF-α与促炎细胞因子的生成,并诱导巨噬细胞活化标志物MHC-II、CD80和CD86的表达,这表明细胞向M1极化;同时降低自噬标志物LC3BII和P62/SQSTM1的水平,增加了P62/Beclin1的比例,使得细胞自噬受到抑制。这些内环境变化都抑制了有效的先天免疫反应, 保证Mtb在巨噬细胞中的存活[47]。此外有报道Mtb-ΔripA未能在巨噬细胞和小鼠模型中有效建立感染,也表明RipA在感染中所起的关键作用[48]。目前还没有相关化合物的筛选报告,但RipA作为抗结核靶点已经展现出其研究前景。
酰胺酶Ami1和Ami4参与细胞分裂过程中的肽聚糖降解、再循环等过程,其中Ami1被认为与RipA共同保障Mtb在宿主细胞内的分裂[48]。Kieswetter等[49]通过在C57BL/6小鼠模型中分别对Ami1和Ami4进行敲除实验,结果显示趋化因子以及促炎细胞因子皆有所提升,然而实验结果并不稳定,后续还需要进一步的探究。
除了上述分类的靶点以外,还有许多在Mtb的生长、致病等过程中起关键作用的蛋白。如与蛋白质合成相关的亮氨酰-tRNA合成酶[50]、与细胞壁合成相关的聚酮合酶[51]及β-酮酰-ACP合成酶[52]、与能量代谢相关的异柠檬酸裂解酶[53]等,都具备成为抗结核药物靶点的潜力。
3 总结
近几十年来随着对Mtb各种代谢相关基因的破解和生物技术的进步,抗结核靶点的研究进展加快,许多有潜力的靶点得以被发现。通过对靶点基因组学和耐药机制的深入探究,又筛选出众多具备潜在抗结核能力的新型化合物。不过由于化合物从筛选、合成到试验的周期长、成本高,加上体内外的实验结果具有差异性,因而目前进入临床试验阶段的化合物并不多,近几十年来上市的新型化合物仍然只有贝达喹啉属于首选药物。尽管如此,针对重要靶点如细胞壁合成的抑制剂SQ109、TBA-7371,能量代谢相关的抑制剂Q203和TBAJ-587等研究已较为成熟且仍在优化试验,有望上市。
本文参考《中国抗生素杂志》2017年“抗结核靶点的研究进展”一文[54],对DprE、Mmp L3等几个之前介绍过的重要靶点进行了最新的综述,并按照影响Mtb的细胞壁合成、DNA/RNA合成、能量代谢等方向,对近年来报道的其它靶点及相关化合物进行归纳分析,同时对于一些目前研究较少但具有潜在作用的如免疫相关靶点也进行了简要综述。总之,对Mtb的生存及其代谢途径进行多方位的研究,有利于开发高效的抗结核药物,有望早日为患者带来福音。
参 考 文 献
[1]WHO | Global tuberculosis report 2021[R]. WHO, 2021.
[2]O'Neill J. Tackling drug-resistant infections globally: Final report and recommendations[J/OL]. http://amr-review.org/sites/default/files/160525_Final%20paper_with%20cover.pdf.
[3]Chakraborty S, Rhee K Y. Tuberculosis drug development: History and evolution of the mechanism-based paradigm[J]. Cold Spring Harb Perspect Med, 2015, 5(8): a021147.
[4]Bahuguna A, Rawat D S. An overview of new antitubercular drugs, drug candidates, and their targets[J]. Med Res Rev, 2020, 40(1): 263-292.
[5]鄧琪, 肖春玲. 新型抗结核药物研究进展[J]. 中国新药杂志, 2019, 28(13): 1567-1573.
[6]Kb S, Kumari A, Shetty D, et al. Structure based pharmacophore modelling approach for the design of azaindole derivatives as DprE1 inhibitors for tuberculosis[J]. J Mol Graph Model, 2020, 101: 107718.
[7]Zhang B, Li J, Yang X, et al. Crystal structures of membrane transporter MmpL3, an anti-TB drug target[J]. Cell, 2019, 176(3): 636-648.
[8]Egbelowo O, Sarathy J P, Gausi K, et al. Pharmacokinetics and target attainment of SQ109 in plasma and human-like tuberculosis lesions in rabbits[J]. Antimicrob Agents Chemother, 2021, 65(9): e0002421.
[9]贾一鹤, 吴梦同, 刘家族, 等. 结核分枝杆菌药物靶点的研究进展[J]. 中南药学, 2018, 16(7): 894-903.
[10]Rath J P, Raval M K. Structure based screening of ligands against dTDP-6-deoxy-D-xylo-4-hexulose 3, 5-epimerase (RmlC): Phytochemical as drug candidate for Mycobacterium tuberculosis[J]. Phar Biol Eval, 2017, 4(2): 97-102.
[11]Dhaked D K, Bala Divya M, Guruprasad L. A structural and functional perspective on the enzymes of Mycobacterium tuberculosis involved in the L-rhamnose biosynthesis pathway[J]. Prog Biophys Mol Biol, 2019, 145: 52-64.
[12]Chollet A, Maveyraud L, Lherbet C, et al. An overview on crystal structures of InhA protein: Apo-form, in complex with its natural ligands and inhibitors[J]. Eur J Med Chem, 2018, 25(146): 318-343.
[13]Armstrong T, Lamont M, Lanne A, et al. Inhibition of Mycobacterium tuberculosis InhA: Design, synthesis and evaluation of new di-triclosan derivatives[J]. Bioorg Med Chem, 2020, 28(22): 115744.
[14]Bruning J B, Murillo A C, Chacon O, et al. Structure of the Mycobacterium tuberculosis D-alanine: D-alanine ligase, a target of the antituberculosis drug D-cycloserine[J]. Antimicrob Agents Chemother, 2011, 55(1): 291-301.
[15]Yang S, Xu Y, Wang Y, et al. The biological properties and potential interacting proteins of D-alanyl-D-alanine ligase A from Mycobacterium tuberculosis[J]. Molecules, 2018, 23(2): 324.
[16]Meng J, Gao P, Wang X, et al. Digging deeper to save the old anti-tuberculosis target: D-alanine-D-alanine ligase with a novel inhibitor, IMB-0283[J]. Front Microbiol, 2020, 10: 3017.
[17]Dubaniewicz A, Holownia A, Kalinowski L, et al. Is mycobacterial heat shock protein 16 kDa, a marker of the dormant stage of Mycobacterium tuberculosis, a sarcoid antigen[J]? Hum Immunol, 2013, 74(1): 45-51.
[18]Moradi B, Sankian M, Amini Y, et al. A new DNA vaccine expressing HspX-PPE44-EsxV fusion antigens of Mycobacterium tuberculosis induced strong immune responses[J]. Iran J Basic Med Sci, 2020, 23(7): 909-914.
[19]Mdluli K, Ma Z. Mycobacterium tuberculosis DNA gyrase as a target for drug discovery[J]. Infect Disord Drug Targets, 2007, 7(2): 159-168.
[20]Chaudhari K, Surana S, Jain P, et al. Mycobacterium tuberculosis (MTB) GyrB inhibitors: An attractive approach for developing novel drugs against TB[J]. Eur J Med Chem, 2016, 124: 160-185.
[21]Gl B, Rajput R, Gupta M, et al. Structure-based drug repurposing to inhibit the DNA gyrase of Mycobacterium tuberculosis[J]. Biochem J, 2020, 477(21): 4167-4190.
[22]Rothstein D M. Rifamycins, alone and in combination[J]. Cold Spring Harb Perspect Med, 2016, 6(7): a027011.
[23]Hameed S, Moganeradj K, Mahmood N, et al. Sequence analysis of the rifampicin resistance determining region (RRDR) of rpoB gene in multidrug resistance confirmed and newly diagnosed tuberculosis patients of Punjab, Pakistan[J]. PLoS One, 2017, 12(8): e0183363.
[24]Zaw M T, Emran N A, Lin Z. Mutations inside rifampicin-resistance determining region of rpoB gene associated with rifampicin-resistance in Mycobacterium tuberculosis[J]. J Infect Public Health, 2018, 11(5): 605-610.
[25]Biazus G, Schneider C Z, Palma M S, et al. Hypoxanthine-guanine phosphoribosyltransferase from Mycobacterium tuberculosis H37Rv: Cloning, expression, and biochemical characterization[J]. Protein Expr Purif, 2009, 66(2): 185-190.
[26]Eng W S, Rejman D, Pohl R, et al. Pyrrolidine nucleoside bisphosphonates as antituberculosis agents targeting hypoxanthine-guanine phosphoribosyltransferase[J]. Eur J Med Chem, 2018, 159: 10-22.
[27]Song L, Merceron R, Gracia B, et al. Structure guided lead generation toward nonchiral M. tuberculosis thymidylate kinase inhibitors[J]. J Med Chem, 2018, 61(7): 2753-2775.
[28]Cui Q, Shin W S, Luo Y, et al. Thymidylate kinase: An old topic brings new perspectives[J]. Curr Med Chem, 2013, 20(10): 1286-1305.
[29]Wayengera M, Kateete D P, Asiimwe B, et al. Mycobacterium tuberculosis thymidylate kinase antigen assays for designating incipient, high-risk latent M. tb infection[J]. BMC Infect Dis, 2018, 18(1): 133.
[30]Naik M, Raichurkar A, Bandodkar B S, et al. Structure guided lead generation for M. tuberculosis thymidylate kinase (Mtb TMK): discovery of 3-cyanopyridone and 1,6-naphthyridin-2-one as potent inhibitors[J]. J Med Chem, 2015, 58(2): 753-766.
[31]Venugopala K N, Tratrat C, Pillay M, et al. In silico design and synthesis of tetrahydropyrimidinones and tetrahydropyrimidinethiones as potential thymidylate kinase inhibitors exerting anti-TB activity against Mycobacterium tuberculosis[J]. Drug Des Devel Ther, 2020, 14: 1027-1039.
[32]Chatterjee A, Pandey S, Dhamija E, et al. ATP synthase, an essential enzyme in growth and multiplication is modulated by protein tyrosine phosphatase in Mycobacterium tuberculosis H37Ra[J]. Biochimie, 2019, 165: 156-160.
[33]Saxena A K, Alam M. ATP synthase inhibitors as anti-tubercular agents: QSAR studies in novel substituted quinolines[J]. Curr Top Med Chem, 2020, 20(29): 2723-2734.
[34]Dhulap A, Banerjee P. ATP synthase, an emerging target in TB drug discovery: Review of SAR and clinical pharmacology of diarylquinoline inhibitors[J]. Curr Drug Targets, 2021, 22(11): 1207-1221.
[35]Cai Y, Jaecklein E, Mackenzie J S, et al. Host immunity increases Mycobacterium tuberculosis reliance on cytochrome bd oxidase[J]. PLoS Pathog, 2021, 17(7): e1008911.
[36]Kalia N P, Hasenoehrl E J, Ab Rahman N B, et al. Exploiting the synthetic lethality between terminal respiratory oxidases to kill Mycobacterium tuberculosis and clear host infection[J]. Proc Natl Acad Sci U S A, 2017, 114(28): 7426-7431.
[37]Cleghorn L A T, Ray P C, Odingo J, et al. Identification of Morpholino Thiophenes as novel Mycobacterium tuberculosis inhibitors, targeting QcrB[J]. J Med Chem, 2018, 61(15): 6592-6608.
[38]Gordon S, Simithy J, Goodwin D C, et al. Selective Mycobacterium tuberculosis shikimate kinase inhibitors as potential antibacterials[J]. Perspect Med Chem, 2015, 7: 9-20.
[39]Masoko P, Mabusa I H, Howard R L. Isolation of alpha-linolenic acid from sutherlandia frutescens and its inhibition of Mycobacterium tuberculosis' shikimate kinase enzyme[J]. BMC Complement Altern Med, 2016, 16: 366.
[40]Rahul Reddy M B, Krishnasamy S K, Kathiravan M K. Identification of novel scaffold using ligand and structure based approach targeting shikimate kinase[J]. Bioorg Chem, 2020, 102: 104083.
[41]Syed P R, Chen W, Nelson D R, et al. Cytochrome P450 monooxygenase CYP139 family involved in the synthesis of secondary metabolites in 824 Mycobacterial species[J]. Int J Mol Sci, 2019, 20(11): 2690.
[42]Sangana R, Gu H, Chun D Y, et al. Evaluation of clinical drug interaction potential of clofazimine using static and dynamic modeling approaches[J]. Drug Metab Dispos, 2018, 46(1): 26-32.
[43]Bukhdruker S, Varaksa T, Grabovec I, et al. Hydroxylation of antitubercular drug candidate, SQ109, by Mycobacterial cytochrome P450[J]. Int J Mol Sci, 2020, 21(20): 7683.
[44]Ferguson L, Wells G, Bhakta S, et al. Integrated target-based and phenotypic screening approaches for the identification of anti-tubercular agents that bind to the Mycobacterial adenylating enzyme MbtA[J]. Chem Med Chem, 2019, 14(19): 1735-1741.
[45]Bythrow G V, Mohandas P, Guney T, et al. Kinetic analyses of the siderophore biosynthesis inhibitor Salicyl-AMS and analogues as MbtA inhibitors and antimycobacterial agents[J]. Biochemistry, 2019, 58(6): 833-847.
[46]Salimizand H, Jamehdar S A, Nik L B, et al. Design of peptides interfering with iron-dependent regulator (IdeR) and evaluation of Mycobacterium tuberculosis growth inhibition[J]. Iran J Basic Med Sci, 2017, 20(6): 722-728.
[47]Shariq M, Quadir N, Sharma N, et al. Mycobacterium tuberculosis RipA dampens TLR4-mediated host protective response using a multi-pronged approach involving autophagy, apoptosis, metabolic repurposing, and immune modulation[J]. Front Immunol, 2021, 12: 636644.
[48]Healy C, Gouzy A, Ehrt S. Peptidoglycan hydrolases RipA and Ami1 are critical for replication and persistence of Mycobacterium tuberculosis in the host[J]. mBio, 2020, 11(2): e03315-e03319.
[49]Kieswetter N S, Ozturk M, Jones S S, et al. Deletion of N-acetylmuramyl-L-alanine amidases alters the host immune response to Mycobacterium tuberculosis infection[J]. Virulence, 2021, 12(1): 1227-1238.
[50]Palencia A, Li X, Bu W, et al. Discovery of novel oral protein synthesis inhibitors of Mycobacterium tuberculosis that target leucyl-tRNA synthetase[J]. Antimicrob Agents Chemother, 2016, 60(10): 6271-6280.
[51]Wang X, Zhao W, Wang B, et al. Identification of inhibitors targeting polyketide synthase 13 of Mycobacterium tuberculosis as antituberculosis drug leads[J]. Bioorg Chem, 2021, 114: 105110.
[52]Brown A K, Sridharan S, Kremer L, et al. Probing the mechanism of the Mycobacterium tuberculosis beta-ketoacyl-acyl carrier protein synthase III mtFabH: Factors influencing catalysis and substrate specificity[J]. J Biol Chem, 2005, 280(37): 32539-32547.
[53]Sharma R, Das O, Damle S G, et al. Isocitrate lyase: A potential target for anti-tubercular drugs[J]. Recent Pat Inflamm Allergy Drug Discov, 2013, 7(2): 114-123.
[54]耿葉慧, 李子强, 张瑜. 抗结核靶点的研究进展[J]. 中国抗生素杂志, 2017, 42(2): 90-97.
