固态电解质中的聚合物复合体系研究进展
2022-05-23董常熠于德梅
董常熠,于德梅
(西安交通大学 化学学院,西安 710049)
要成功从濒临枯竭的化石燃料过渡到可再生能源,关键在于找到高效、安全和环境友好的储能方式。1859年,法国物理学家加斯顿-普兰特成功制备了铅酸蓄电池,极大地推动了电源的便携化、小型化,从此可充电电池开始了蓬勃发展[1]。如今,各类电子设备中采用的充电电池主要是锂离子电池。其通常以石墨作为负极,以LixCoO2,LixNiO2,LixMnO4等为正极,利用特定的有机电解液实现锂离子在其中的迁移和在电极材料上的嵌入/脱嵌[2]。近年来,超级电容器由于其优越的功率密度和较长的循环寿命,也作为一类重要的储能系统受到科研人员的广泛关注[3]。
电解质是这些储能设备中的重要组成部分。然而,目前商业应用最广也最成熟的有机液态电解质高度易燃,实际使用中具有较大的安全隐患;而且在发生泄漏后会释放有毒的电解液,易对环境产生较大损害;特别是有机电解液和新一代高能量密度材料兼容性差的缺点,严重限制了储能器件能量密度的提升[4-6]。近年来,可穿戴电子设备的飞速发展,更要求储能器件具有在复杂变形条件下正常工作的能力,传统的有机电解液即使进行严密的封装以避免泄露,也难以达到所需的柔韧性和延展性[7-8]。因此固态电解质是解决上述问题,开发下一代高比能量、高柔韧性、高安全性电化学器件的关键选择。
根据主体材料的不同,固态电解质可分为两大类:固体聚合物电解质(SPE) 和无机固态电解质(ISE)。要实现固态电解质的商业应用,必须同时确保: (1)足够的离子导电能力(25 ℃下大于10-4S·cm-1); (2)足够高的机械强度;(3)至少4~5 V的电化学稳定窗口[9]。无机固态电解质主要包括Li1+xAlxTi2-x(PO4)3(LATP),Li7La3Zr2O12(LLZO) 等,往往表现出较高的化学和电化学稳定性、机械强度和离子电导率,但其与电极材料接触性能差,易造成较大的界面阻抗[10]。固态聚合物电解质质量轻、柔性好,且与电极材料接触良好、界面阻抗小,但是纯固态聚合物电化学窗口较窄、力学性能较差、离子电导率较低(10-5S·cm-1以下)的缺点限制了其实际应用。
固态聚合物电解质和无机填料结合可以有效提高离子电导率并增强主体的力学性能,达到防止泄露和阻碍枝晶生长的作用[11-12]。除此以外,聚合物电解质本身还可采取共聚[13-14]、交联[15-16]、共混[6,17-18]等多种方法增强性能,满足实际需要。本文从聚合物研究的角度出发,结合本课题组前期积累的工作基础,专注于固态聚合物在电解质中的应用和改性策略,对复合固态聚合物电解质(CPE) 近年来的研究进展进行了系统的总结和评述。
1 固态聚合物电解质导电机理

1.1 晶体空位扩散模型
该模型主要研究离子在聚合物结晶区的离子迁移行为,聚合物链在结晶区形成如图1(a)所示的螺旋形构象。在电场作用下,电解质盐解离形成的阳离子在螺旋体孔道内进行定向迁移,而阴离子在孔道外螺旋结构之间反向迁移。显然,阳离子的迁移速度受阴阳离子作用力的影响,当阴阳离子作用较强时,离子迁移行为受阻,离子电导率降低。不同的聚合物在不同的温度下将会形成不同的螺旋构象,进而导致孔道内外阴阳离子作用力的不同,影响离子电导率。在这种传导机理中,离子电导率和温度的关系满足Arrhenius方程(式(1))。
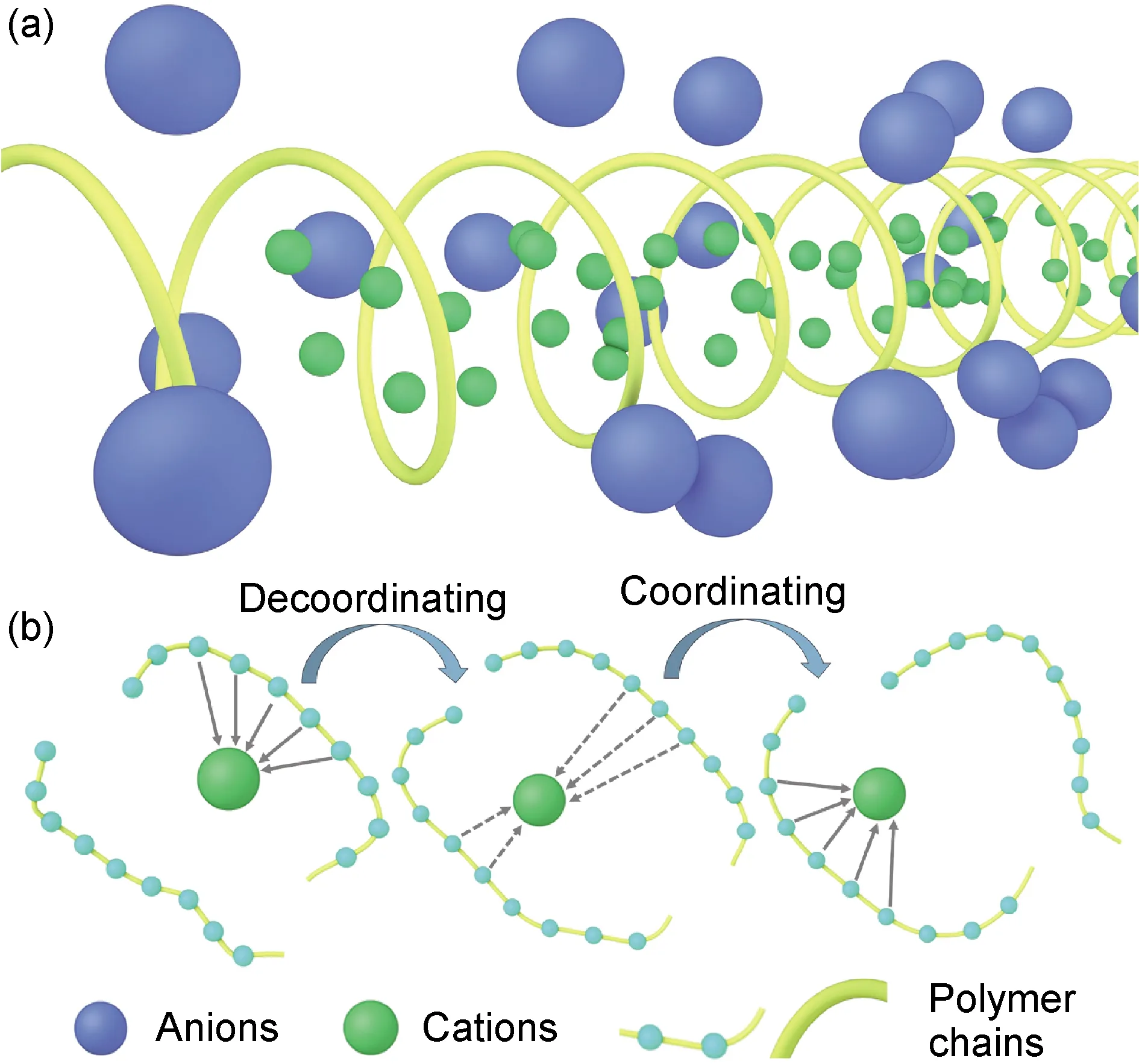
图1 固态聚合物电解质导电机理示意(a)晶体空位扩散模型;(b)非晶区扩散模型Fig.1 Conductive mechanism of solid polymer electrolytes(a)crystal vacancy diffusion model;(b)amorphous region diffusion model
(1)
式中:σ为离子电导率;A为指前因子;T为环境温度;Ea为离子传导的表观活化能;R为理想常数。然而,离子在结晶区的迁移速率往往较小,且许多聚合物在常温下处于半晶态或非晶态,因此事实上大部分的离子电导率由非晶区扩散贡献,这就提出了非晶区扩散模型。
1.2 非晶区扩散模型
当温度升高至玻璃化转变温度以上时,聚合物进入高弹态,部分分子链从高度取向的玻璃态解取向出来,链段运动能力提高。此时,离子在其中的迁移可以用如图1(b)所示的非晶区扩散模型解释。首先借助聚合物链段上某些具有Lewis碱特性的原子与电解质盐的阳离子配位,增大盐类解离的程度;然后随着聚合物链段的运动和电场的作用,阳离子在不同链段之间发生不断地配位-解配位-配位,实现它在聚合物内部的连续迁移。在这种传导机理中,离子电导率和温度的关系满足VTF (Vogel-Tamman-Fulcher) 方程(式(2))。
(2)
式中:K为常数;T0为动力学理想玻璃化转变温度,在此温度下聚合物由构象变化导致的熵变为0,通常取T0=Tg-50 K[20];Tg为聚合物的玻璃化转变温度;其余符号σ,A,T,Ea的意义与式(1)相同。
因此传统观念认为,尽可能选用Tg较低的聚合物作为电解质基体有助于其在常温下表现出较高的链段运动能力,促进离子迁移,提高离子电导率。但Zhang等[21]的研究也表明,聚碳酸丙烯酯尽管具有较高的Tg(4.8 ℃),同样可以展现出优异的离子电导率。可见SPE的离子电导率不仅取决于Tg,同时还受多种因素的影响,未来提升电导率的策略不应仅局限于选取Tg更低的材料。
1.3 自由体积模型
自由体积模型为离子在聚合物中扩散具体受哪些因素的影响做了进一步的解释。该模型认为,聚合物中的分子链会发生一定程度的振动,这种振动将形成分子周围空间的大小变化,这个空间就是自由体积。自由体积的大小随着分子振动能量的升高而增大,当自由体积的大小大于电解质盐解离产生的离子的体积时,这些离子就会随着自由体积大小和位置的变化而迁移。由自由体积理论可以推导出WLF(Williams-Landel-Ferry) 方程(式(3)),描述聚合物链段松弛时间和温度的关系。
(3)
式中:Tr为参考温度,通常用Tg表示;aT为温度为T和Tg时链段松弛时间的比值,当聚合物密度随温度变化不大时,可以用聚合物在不同温度下的黏度之比表示;C1和C2为常数,与Tr取值有关,当Tr用Tg表示时,C1和C2分别为17.44和51.60。
除上述三种应用范围较广的模型以外,MacGlashan等[22]也曾提出,在聚氧乙烯(PEO) 基中,当EO∶Li+(摩尔比)为6∶1时,往往展现出与上述模型均不同的离子导电机理。两条PEO链采用非螺旋构象合并组成一个圆筒状结构,Li+在圆筒中与一条链上的两个醚氧以及另一条链上的三个醚氧分别配位,形成5个配位键,而电解质盐阴离子在圆筒结构的外部,不与Li+存在配位作用。这就使得电解质盐的阴阳离子可以在圆筒结构的内外两侧分别迁移、不受干扰,极大地提升了离子电导率。这一研究证明除了温度以外,盐浓度也会影响聚合物链结构进而改变离子传输机理。
总之,聚合物中离子传导机理非常复杂,有时还会同时具有上述多种机理,因此未来还需要研究人员的深入研究,提出更具有普适性和更准确的模型。
2 固态聚合物复合体系
为了更好地介绍不同聚合物在复合固态电解质中的应用,本文以复合体系基体所用聚合物的不同进行了归纳。以导电机理进行归纳,这些聚合物被归纳为多离子导电机理和单离子导电机理;基于介电常数的大小,分为低介电常数聚合物和高介电常数聚合物。由于大部分的多离子导电聚合物都为低介电常数,因此本节将这些聚合物归纳为三类:多离子导电聚合物复合体系、单离子导电聚合物复合体系和高介电常数聚合物复合体系。
2.1 多离子导电聚合物复合体系
多离子导电聚合物复合体系的研究始于1973年,Wright等[23]对聚氧乙烯(PEO) 与碱金属离子络合物导电性的研究发现,至今该体系已经成为研究时间最长、研究范围最广的聚合物电解质体系。其基本组成为聚合物基体以及电解质盐,电解质盐在聚合物中解离为阴离子和阳离子,在电场作用下分别向两电极迁移,从而构成电化学器件的内电路,与电子迁移构成的外电路一起,形成完整的回路。但是由于聚合物固有的结晶度、链段运动能力等方面的限制,离子在聚合物基体中的迁移往往并不容易,同时力学性能、热稳定性等也是聚合物电解质需要考虑的因素,这些方面的缺陷都极大地影响了电化学器件的性能提升[5]。近年来研究人员采用多种方式对已有的聚合物进行改性,并设计路线合成新的具有特定功能的聚合物,表1[10,15,18,24-36]列出了一些聚合物的改性策略和对性能的影响。本文选取应用前景较广的聚合物基体,将多离子导电聚合物体系分为聚氧乙烯(PEO) 基、聚丙烯腈(PAN) 基和聚乙烯醇(PVA) 基,并在最后对其他尚未提及但仍有亮点的聚合物体系进行了综述。

表1 一些聚合物的改性策略及其性能表征结果Table 1 Modification strategies and characterization results of some polymers
2.1.1 聚氧乙烯(PEO) 基聚合物电解质
聚氧乙烯(PEO) 被广泛认为是高能量密度二次锂电池固态电解质中最有前途的聚合物基质候选材料,也是目前为止研究最广泛的聚合物电解质基底。受到其氧原子上孤对电子影响,PEO聚合物链往往可以通过与金属离子发生配位作用而展现出一定的电导率。20世纪80年代起,美国西北大学的Shriver课题组率先对PEO基复合电解质的离子传输机理、离子迁移数、光谱数据等展开了一系列基础研究[37-42],为后来相关科研项目的开展奠定了坚实的基础。1983年,Armand等[4,43]分别研究了PEO和锂盐与钠盐复合物,并提出将PEO作为未来聚合物电解质开发的基底材料。PEO基质的优势主要表现在:(a) 分子链中醚氧键含量高且柔顺性好,因此更易与金属离子配位,具有良好的相容性[44-45];(b) 具有较好的电化学稳定性,在常见工作环境下性能更加稳定[46]。但由于介电常数较低(ε≈8),PEO往往展现出较严重的离子聚集现象[47],再加上其室温下易于结晶的性质[48],使得离子在聚合物分子链中迁移的难度大大提高,室温离子电导率较低(约为10-6~10-5S·cm-1)[7,49]。如何在保证稳定性和力学性能的前提之下提高PEO的离子电导率成为当前研究的热点问题。
1991年,Capuano等[11]在添加β-Al2O3粉末提升PEO固态电解质力学性能的过程中发现,当陶瓷微粉的粒径小于5 μm时,电解质的电化学性能也将得到显著提高。这是因为分散无机粒子一方面影响PEO聚合物链的结晶动力学,从而促进局部非晶区的形成,最终增强Li+的传输;另一方面也在纳米颗粒和聚合物之间形成新的相界面,由于陶瓷颗粒表面有大量的空隙可以容纳电解质离子,从而实现离子在颗粒之间的活性跳跃移动[50]。Wu等[10]使用LiZr2(PO4)3(LZP) 作为PEO电解质中的填料,研究发现,LZP颗粒表面与聚合物基质之间的相互作用增加了Li+的迁移数,并使Li+在两种不同的局部环境中重新分布。很大一部分Li+被重新分配到无序的局部环境,这为Li+提供了更大的迁移数,并改善了聚合物复合材料的整体导电性。吴浩等[51]通过溶胶-凝胶法制备得到水铝英石,与LiClO4一同共混加入PEO制备成CPE,发现水铝英石中的—OH可以借助氢键作用降低PEO的结晶度,此外水铝英石与锂盐的相互作用也可以促进锂盐的解离,借助Li+与聚合物链段的络合作用也可以进一步抑制PEO结晶。当填料达到最佳配比时,PEO结晶度仅为4.12%,球晶最大尺寸仅为12 μm。但Verma等[52]也证明无机填料含量过高将导致离子聚集现象,从而降低固态电解质的电导率,因此对不同的聚合物基质和无机填料,选用不同的浓度配比将十分重要。
研究表明,无机粒子的比表面积越大,对聚合物离子导电性的改善作用越强。为进一步增大无机离子的比表面积,近年来金属有机骨架(MOF) 作为一种由金属原子和有机配体周期性有序形成的新型多孔晶体材料正逐渐引起关注。这类材料具有高比表面积,在同样的体积内能够负载更多的带电物质,有助于离子在其内部的快速跳跃迁移、降低离子传输的能垒,从而改善电解质的离子电导率。Wu等[53]开发了一种铈基MOF填料。三维结构的Ce-MOF颗粒作为交联中心分布在PEO基体中,其内部丰富的Lewis酸性位点对PEO中的氧原子和锂盐阴离子展现出强烈的相互作用,不仅降低了聚合物结晶度,还提高了锂离子迁移数(0.75) 和电导率(30 ℃时为3.0×10-5S·cm-1),同时韧性提高到了未加Ce-MOF时的两倍。
Yang等[54]基于核磁共振波谱测试(NMR)发现,一维纳米纤维的嵌入可以部分修饰聚合物基体,为锂离子传导创造更快的路径,从而使得其对离子电导率的提升效果优于掺杂陶瓷颗粒。为进一步利用无机陶瓷颗粒离子电导率高的优势,近年来研究人员还尝试制备连续相的陶瓷纳米结构并嵌入聚合物电解质中。Wang等[55]采用冰模板法(ice-templated) 将Li1.5Al0.5Ge1.5(PO4)3(LAGP) 纳米颗粒构筑成二维结构上竖直排列的“墙”,再用PEO浇铸,形成的复合电解质室温下电导率可达1.67×10-4S·cm-1,是LAGP颗粒在聚合物中随机分散时的6.9倍。
除了掺入无机粒子,当PEO与其他聚合物共混时同样可以有效抑制结晶、提升离子电导率。徐玲[6]利用溶液浇筑法制备了PEO-PVA-PESf共混体系的固态电解质,由于PVA和PESf对共混体系的协同作用促进了盐离子的非晶化和解离,使得复合电解质展现出5.38 V的宽电压窗口和60 ℃下0.83×10-3S·cm-1的高离子电导率。
通过紫外光辐射等方法还可以使聚合物发生交联,交联使PEO具有类似橡胶的质地和大范围的无定形相,这种无定形相增强了聚合物的离子导电性,并可以增强力学性能,防止材料的蠕变行为发生[56-57]。Wang等[58]以三甲氧基硅基封端的聚丙二醇(SPPG) 作为聚合物基体,以双草酸硼酸锂(LiBOB) 作为锂盐,同时利用其中的BOB-作为SPPG的交联点,构建交联聚合物的同时还限制了阴离子的迁移,将锂离子迁移数提高到0.65,室温下离子电导率提高到10-4S·cm-1量级,特别是在电极表面原位聚合生成电解质后电池器件展现出了优良的循环性能,证明该电解质在高能量密度的锂离子电池中具有广阔的应用前景。
在分子链结构的层面,可以通过共聚或接枝改善分子链的运动能力和Li+的迁移能力,从而降低聚合物的结晶度、提升Li+的传输性能[48]。Petrov等[13]制备的环氧乙烷(EO) 和环氧丙烷(PO) 共聚物中,当PO的摩尔分数达到44%时,共聚物的结晶度仅为同等条件下PEO均聚物的10%。
2.1.2 聚丙烯腈(PAN) 基聚合物电解质
聚丙烯腈(PAN) 有着优良的耐热性、阻燃性和稳定性,这使得它能够展现出极高的安全性,同时其制备成本低、电压窗口宽的特点也使之成为未来高电压电池的理想电解质材料[59]。相对于PEO,由于PAN中不含氧原子,其中的氮原子又和锂离子之间的相互作用力较低,因此锂离子往往可以在PAN电解质中达到比较均匀的分散,迁移数tLi+可达0.5左右,因此这一聚合物近年来成为锂离子电池研究人员关注的重点。
Li等[60]研究了Li1.3Al0.3Ti1.7(PO4)3(LATP) 的掺杂量对PAN离子电导率的影响,他们发现,当LATP掺杂量为15%(质量分数,下同)时,PAN/LATP/LiClO4复合电解质离子电导率达到最大值,室温下为2.14×10-5S·cm-1。同时LATP的加入还使PAN/LiClO4体系的电压窗口从3.7 V升高到4.5 V以上,拉伸强度从4.64 MPa升高到5.34 MPa。但是显然仅仅添加无机纳米颗粒对于复合电解质的离子电导率提升有限,因此,Hu等[61]制备了表面多孔的Li0.33La0.557TiO3(LLTO) 纳米管并作为填料加入PAN中。与一般的纳米颗粒和纳米线不同,他们制备的多孔纳米管与聚合物基体之间接触润湿性更好,对电解质阴离子吸附作用更强,同时大的比表面积也为离子传输提供了更大规模的导电区。该CPE室温离子电导率达到3.6×10-4S·cm-1,电化学窗口提高到5 V,特别是由其组装成的Li-Li对称电池稳定工作时间达到1000 h以上,极化电压只有0.02 V,而裸PAN电解质的极化电压则超过0.5 V,稳定工作时长不到200 h。
由于PAN本身的离子电导率较低,研究人员常常向其中加入有机溶剂作为增塑剂,使聚合物发生溶胀制成凝胶型聚合物电解质。例如,当在130~150 ℃时添加碳酸丙烯酯(PC) 配制成20%的PAN溶液后,将溶液冷却,即可得到凝胶态的PAN[62]。Huang等[63]通过红外光谱和拉曼光谱证实了聚丙烯腈、增塑剂和锂盐之间存在相互作用,正是这种相互作用影响了Li+在电解质中的解离与配位。因此这样的凝胶态PAN可以很轻易地将聚合物电解质的离子电导率提升至10-3S·cm-1级别。常添加的有机溶剂包括碳酸丙烯酯(PC)[27,64]、碳酸乙烯酯(EC)[65-67]、二甲基甲酰胺(DMF)[68]和碳酸二甲酯(DMC)[69]等,为了最大化增塑剂的使用效果,实际应用中往往会配制多种增塑剂的混合体系用于制备凝胶电解质。Saidi等[70]以PAN为主体聚合物,碳酸亚乙酯/碳酸亚丙酯(EC/PC)体系为增塑剂制备了凝胶聚合物电解质,其离子电导率可达8.7×10-3S·cm-1。Verdier等[71]探讨了DMC,PC和二甲基亚砜(DMSO) 三种含有不同浓度锂盐的有机溶剂对PAN基凝胶聚合物离子电导率的影响,他们发现含2 mol/L LiTFSi的PC溶液和质量分数为50%丙烯腈组成的电解质性能最好,室温下离子电导率可达2.1×10-3S·cm-1。
尽管增塑剂的加入使凝胶型电解质的离子电导率大幅提升,但是随着增塑剂含量的升高,凝胶柔性也会上升,力学性能逐渐变差[72-73],这极大限制了PAN基凝胶电解质的实际应用。因此近年来研究人员逐渐把研究重点从离子电导率的提升转移到力学性能的提升上来。目前主要采取的方法是将PAN制成纳米纤维,利用PAN本身力学性能和迁移Li+能力强的优点,作为填料增强整个固态复合电解质的力学性能。Li等[74]首先合成出纳米粒径的LATP颗粒,然后将其与PAN混合溶于DMF,静电纺丝成具有3D结构的骨架,然后将PEO和LiTFSI的混合溶液浇铸在上述骨架上,形成固态复合电解质。测试表明,PAN/LATP纳米纤维的加入使抗拉强度提高了10.72 MPa,60 ℃下复合电解质的离子电导率可达6.5×10-4S·cm-1。Xu等[75]也用类似的方法制备出PAN/LLZO纳米纤维,以EC和碳酸二乙酯(DEC) 为增塑剂,将纳米纤维通过原位聚合法制备得到仅有25 μm厚的凝胶聚合物,这种薄膜电解质具有三条离子传输通道:PAN骨架、LLZO纳米颗粒和连续相的凝胶,因此展现出较高的离子电导率(25 ℃下1.60×10-3S·cm-1)。而3D骨架结构和原位聚合分别保证了薄膜具有足够的机械强度和凝胶与纤维之间良好的界面性能,使得电解质在350 h内进行了稳定的循环过程。
当把PAN基复合电解质应用于锂电池中时,还会存在电极与电解质界面稳定性较差的问题。一方面是因为PAN中的—CN与锂金属之间反应产生钝化层,增大了电池内阻[72],另一方面是因为PAN的刚性较大,与电极的界面接触性能较差[76]。Pan等[77]的研究证明,与电池循环寿命相关的主要是锂金属和聚合物电解质界面的离子电导率,这与电极和电解质的接触性能和生成钝化层的离子电导率息息相关。因此目前的解决方案分为两类:(1) 对电解质的结构进行设计,将PAN设计在内部,为复合电解质整体提供力学性能,而选用其他材料覆盖在PAN外,与锂电极直接接触,确保不生成钝化层或生成的钝化层离子电导率高;(2) 对PAN进行改性,使得电解质整体呈现较好的柔性和较大的比表面积,增强其与电极的接触性能。
Wang等[78]设计了如图2所示的双层电解质,紧贴阳极的为PEO/PAN/LATP复合电解质,其中PEO中的O原子可以与PAN中的氰基形成氢键,削弱其对Li阳极的钝化作用;紧贴阴极的为PAN/LATP复合电解质,其目的在于通过掺杂无机纳米粒子增强电解质整体的离子电导率。测试表明,PAN/LATP复合电解质组成的电池在仅仅40个循环后容量衰减率就达到75%,而与PEO复合后容量衰减仅有不到5%。

图2 双层电解质固态电池整体设计图[78]Fig.2 Structure of solid-state battery with double-layer electrolyte[78]
Liu等[79]通过向PAN凝胶电解质中掺杂笼型聚倍半硅氧烷(POSS) 制备了多孔的凝胶电解质,并制备成Li对称电池进行研究,他们发现掺杂后的电解质表现出良好的界面相容性。如图3所示,当POSS的掺杂量为8%时,随着存放时间的延长,对称电池的钝化层电阻Rp和电荷转移电阻Rct增大速率均远小于未进行掺杂的电池,这表明POSS的掺杂增强了PAN聚合物的柔性,提升了电极与电解质的接触性能。
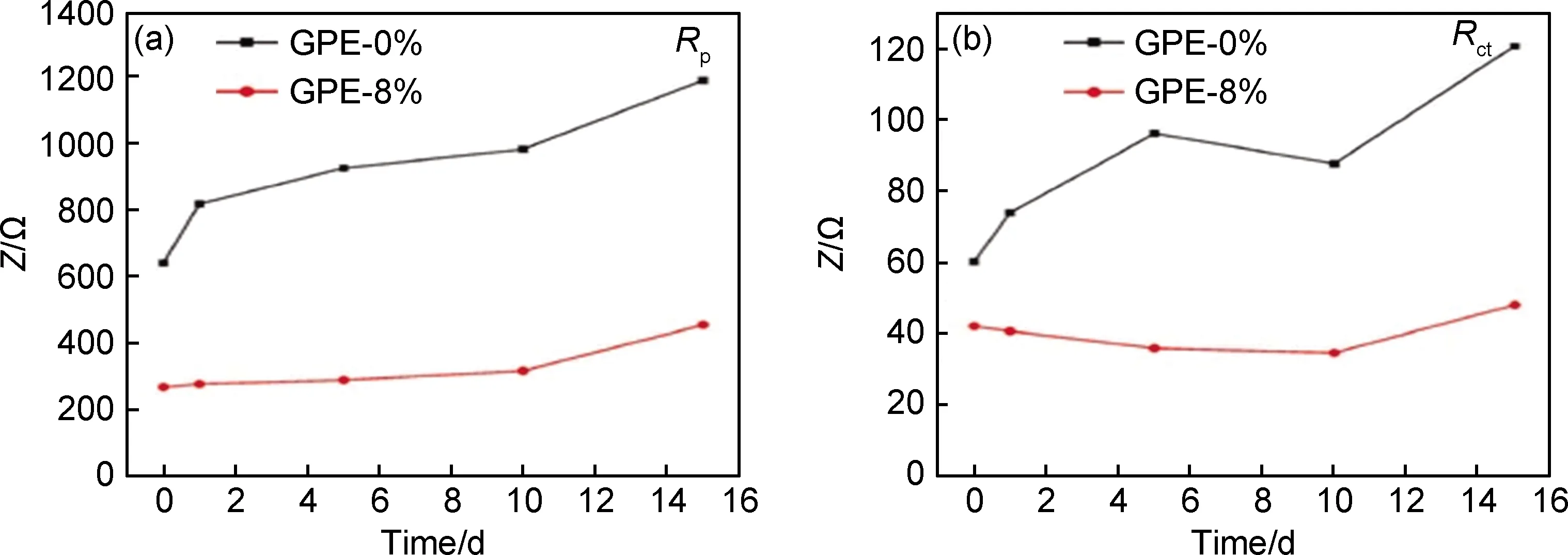
图3 凝胶电解质Rp(a)和Rct(b)随时间变化关系图[79]Fig.3 Change plots of Rp (a) and Rct (b) of GPEs with the storage time[79]
相较于PEO,PAN展现出更宽的电化学窗口和更高的热稳定性,因此在未来高压、高安全性电化学器件中具有更广阔的应用前景。然而PAN结晶性强的特性阻碍了其离子电导率的进一步提升。特别是制备成凝胶后,离子电导率和力学性能这一对矛盾如何同时满足要求,仍然需要进一步的研究与改进。PAN差的力学性能不仅表现在强度难以满足要求上,还表现在其与电极的接触性能不好,再加上—CN与锂电极的“钝化效应”,更使得PAN基CPE的结构需要进行更精妙的设计。但同时也要看到,数十年来PAN在静电纺丝领域获得了大量的研究,许多研究人员结合静电纺丝技术制备了不同形貌和不同负载的PAN纳米纤维。这些纳米纤维可以作为导电网络,也可以作为支撑骨架应用于CPE的制备,将极大地拓宽CPE结构设计的边界。
2.1.3 聚乙烯醇(PVA) 基聚合物电解质
受到内部丰富的羟基的影响,聚乙烯醇通常与水有强烈的相互作用,展现出良好的亲水性[80]。同时其优良的成膜性使其易于制备具有稳定尺寸的电解质薄膜。研究人员结合聚乙烯醇的这两大优点,将其在酸或碱的水溶液中溶胀后制备成聚合物水凝胶电解质。再加上PVA具有价格低廉、生物可降解等优势,近年来逐渐成为固态聚合物电解质研究的热点领域。事实上,借助共混[81-82]、共聚[83-84]和无机粒子掺杂[85-86]等手段,研究人员很早就可以轻易地将PVA基凝胶电解质室温下电解质的离子电导率提升到10-1~10-2S·cm-1级别,因此目前对其的研究更加集中于对实用化方向的探讨,例如碱性镍氢电池、燃料电池等。本文根据凝胶中包含溶液酸碱性的不同,常将PVA基凝胶电解质分为碱性PVA凝胶电解质、酸性PVA凝胶电解质和中性PVA凝胶电解质[87]。
Yang等[80]制备了碱性PVA凝胶电解质并考察了碱盐的不同种类及含量对离子传输性质的影响。如图4(a)所示,碱性PVA-KOH凝胶电解质的离子迁移数最高,且随着浓度的升高,离子迁移数先升高后减小。因此目前PVA基碱性凝胶电解质通常首选的是PVA/KOH体系。Mohamad等[88-89]研究发现,当KOH的质量分数为40%时,PVA/KOH凝胶的电导率最高,达到10-3S·cm-1级别。XRD谱图证明,KOH的加入破坏了PVA晶相,降低了其结晶度,当KOH质量分数达到40%时,PVA晶体衍射峰几乎完全消失,离子电导率达到最大;继续提高KOH的浓度,XRD谱图中将出现未解离的KOH晶体衍射峰,这也是凝胶电导率降低的原因。Tran等[90]证明在锌-空气电池中,相较于传统的KOH溶液电解质,PVA基凝胶态的KOH电解质有助于降低电极-电解质界面电阻和电荷转移电阻。Yang等[16]采用静电纺丝法制备出聚乙烯醇/壳聚糖复合纳米纤维膜,使用戊二醛交联以后浸渍于聚乙烯醇/壳聚糖共混液中,真空干燥后再在40%的KOH溶液中进行长时间浸泡,得到碱性凝胶电解质膜。经过对比发现,这种凝胶电解质膜的室温离子电导率可达0.01 S·cm-1,同时对甲醇有更低的透过率和更高的选择性,将有望用于甲醇燃料电池的聚合物固态电解质中。
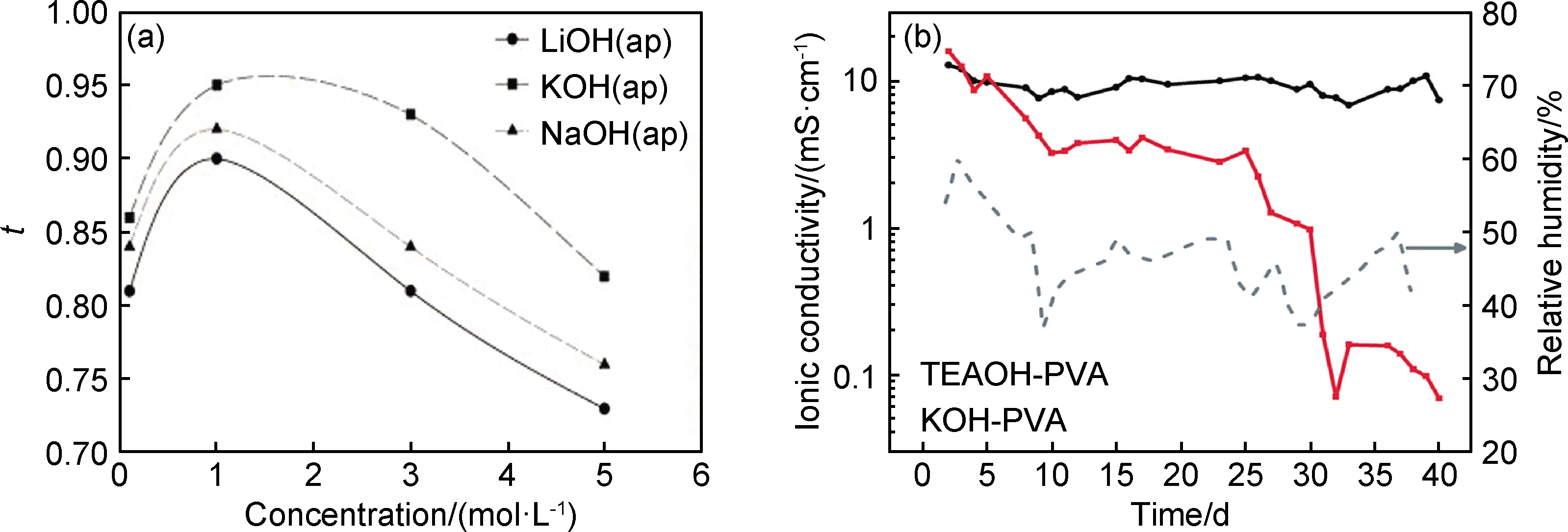
图4 不同种类碱盐对PVA凝胶电解质离子传输性能的影响(a)PVA凝胶中盐的种类与浓度变化对离子电导率的影响[80];(b)KOH-PVA凝胶和TEAOH-PVA凝胶离子电导率随时间的变化关系[87]Fig.4 Effects of different kinds of alkali salts on ion transport properties of PVA gel electrolytes(a)effect of types and concentration changes in PVA gels on ion conductivity[80];(b)ionic conductivity of polymer electrolyte KOH-PVA and TEAOH-PVA over time[87]
然而在实际应用中KOH-PVA体系的水凝胶电解质常常由于保水能力差、KOH易结晶等问题影响器件的寿命和工作稳定性。而季铵盐的引入可以增强电解质与水的作用力,并降低结晶性。多伦多大学Lian团队[87, 91]开发了一种以四乙基氢氧化铵(TEAOH) 替代KOH提供OH-的PVA基碱性凝胶电解质。如图4(b)所示,室温下存放40天后KOH-PVA凝胶脱水严重,KOH大量结晶,电解质离子电导率降低到原来的1%以下,而摩尔比相同的TEAOH-PVA凝胶电解质由于具有优秀的保水能力,离子电导率仍然保持稳定。Li等[92]将这种凝胶电解质运用于柔性锌-空气电池中,相对于传统的KOH-PVA体系,展现出了极好的长期工作的稳定性。如图5放电曲线所示,无论是新制凝胶还是存放5,10,15天以后的凝胶,运用TEAOH-PVA体系的放电时间远超KOH-PVA体系。在此基础上,You等[93]进一步将季铵盐接枝到纤维素上,引入PVA/KOH体系中,其中季铵盐的加入提升了凝胶的保水能力和离子电导率,而纤维素的加入增强了力学性能,使拉伸应力提升了两倍,同时有助于提高凝胶的耐碱性能,延长其在强碱性环境下稳定工作的时间。
图5 TEAOH-PVA凝胶及KOH-PVA凝胶存放不同时间后的放电曲线对比[92](a)新制;(b)5天后;(c)10天后;(d)15天后Fig.5 Comparison of the discharge curve of TEAOH-PVA gel and KOH-PVA gel at different storage time[92](a)fresh state;(b)after 5 days;(c)after 10 days;(d)after 15 days
将质子酸溶液作为水凝胶中的扩散介质时,凝胶电解质的导电机理转变为质子导电,这种凝胶电解质被称为酸性凝胶电解质,由于可以给电容器提供更好的储能特性,近年来常常和石墨烯、碳布以及各种复合材料结合,广泛用于固态超级电容器的研究。水凝胶基超级电容器的电容性能与PVA的浓度密切相关,PVA浓度低,则羟基与羟基之间、羟基与水分子之间距离较远,氢键作用力弱,难以形成稳定的凝胶网络,不利于形成离子传输通道;而PVA浓度过高则会使聚合物链间发生缠结,凝胶网络密度增大,阻碍离子迁移[94]。Ma等[3]以金属氧化物纳米棒/还原氧化石墨烯(rGO) 杂化纤维作为电极,在其上涂覆PVA/H3PO4凝胶作为电解质,制成了纤维状的柔性超级电容器。Wang等[95]制备了超分子碳纳米管(CNF)/聚苯胺(PANI) 气凝胶电极,以PVA/H2SO4凝胶作为电解质,通过0.1 MPa下的压制将其组装成柔性超级电容器,该电容器具有高达291.01 F/g的归一化电容。
双电层电容器(EDLC) 通过电极/电解质界面电荷的静电相互作用储存电能,然而由于电解质的离子扩散效率低,水凝胶基EDLC的电容性能往往较差。Aljafari等[96]研究发现当酸性PVA基凝胶电解质与大孔径碳纳米管电极结合时,会在凝胶体相中产生部分赝电容效应,提升电容器的储能性能,且器件电容与凝胶体积成正比,其氧化还原储能机理如图6所示。但值得注意的是,当电极换成高密度纳米孔径的电极时,流动性更好的液态物质产生的双电层效应将起到主导作用,赝电容效应被削弱。为了进一步提高储能效率,人们通过将无机或有机氧化还原添加剂负载到凝胶基体中,在电解质和电极界面引入可逆的氧化还原过程,开发出氧化还原介导型水凝胶电解质。杂多酸作为一种过渡金属氧化物簇合物因其可以通过快速、可逆的多电子转移过程进行氧化还原反应,近年来获得了广泛的关注。Xie等[97]向PVA-H3PO4凝胶体系中引入磷钼酸PMo12,以碳纸作电极,循环伏安测试证明在电解质/电极界面上发生了氧化还原反应,这使得该超级电容器同时具备了双电层电容和法拉第电容的性质,具有更高的电容性质。充放电测试显示,PMo12使得电容器拥有更长的放电时间和更加对称的充放电曲线,当PMo12/H3PO4质量比为0.5时,比电容相较于未引入PMo12提升了1倍以上。然而尽管如此,其在2000周次循环后容量保持率仅为50%,因此循环稳定性仍需提高。除杂多酸以外,I-/I2离子对、对苯二胺、对苯二酚、亚甲蓝及对苯二酚等也常常作为电解液的添加剂以提升电容性能。
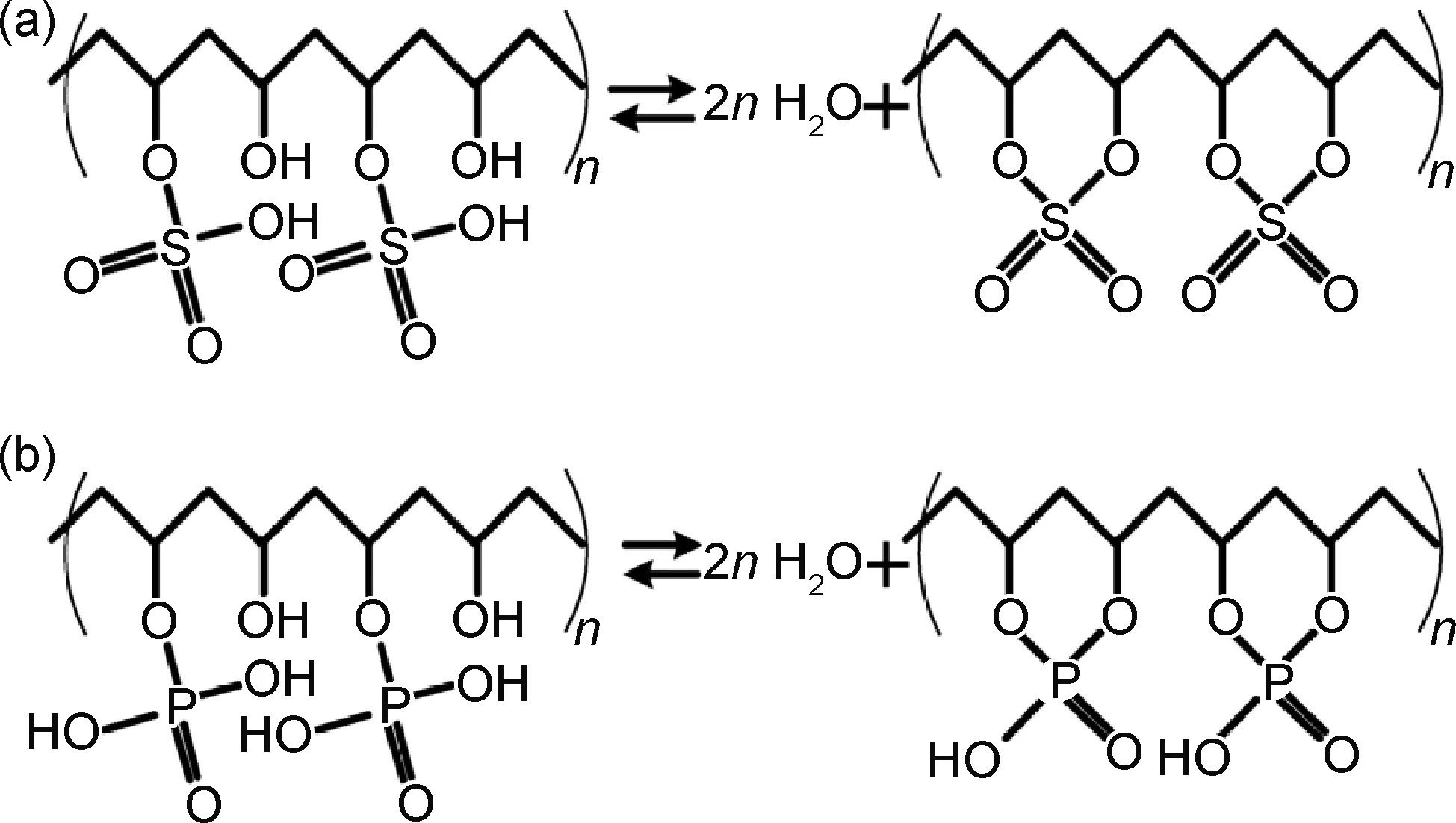
图6 两种酸性PVA凝胶电解质中的电荷储存机理[96](a)PVA-H2SO4凝胶电解质;(b)PVA-H3PO4凝胶电解质Fig.6 Charge storage mechanism in two acid-PVA gel electrolytes[96](a)PVA-H2SO4 gel electrolyte;(b)PVA-H3PO4 gel electrolyte
对于强酸性及强碱性水凝胶电解质,存在以下缺点:(1) 溶液泄漏后容易引发安全问题; (2) 强酸碱性溶液可能会腐蚀集流体,造成器件的结构不稳定,性能衰减; (3) 对于某些赝电容电极材料,例如钒氧化物,在酸性及碱性溶液中溶解度较大,形貌和结构易被破坏,造成大量的容量损失[98]。研究人员尝试在凝胶中加入中性无机盐的水溶液以克服这些缺点。巫梦丹等[99]以碳纳米管阵列(CNTA) 为电极,分别以PVA-NaCl,PVA-CH3COONa,PVA-LiCl 中性凝胶作为电解质制备了超级电容器。测试发现,PVA-NaCl凝胶电解质组成的超级电容器性能最好,最大能量密度可达0.0093 mWh·cm-3,PVA-CH3COONa凝胶电解质组成的器件性能最差,最大能量密度仅为0.0084 mWh·cm-3,但其仍然远高于文献中记载的以PVA-H2SO4凝胶为电解质制备的多壁碳纳米管柔性超级电容器(0.008 mWh·cm-3)。Asim等[100]制备了PVA-LiCl凝胶电解质,并应用于他们设计的电极体系中组成柔性超级电容器,实现了2.35 V的电压窗口和2.5 mA·cm-2时高达184 F·cm-2的电容。
2.1.4 其他种类离子导电聚合物基电解质
相较于无机固态电解质,全固态聚合物电解质具有可加工性能更好、界面相容性更高和更能够适应充放电过程中电极发生的体积变化等优点,其可以被看作是一种聚合物体系中的无溶剂盐溶液。当前科研人员对于全固态聚合物电解质已经进行了大量的研究,单独研究单一聚合物基底带来的性能提升十分有限,必须结合掺杂、共混、共聚等手段并进行适当的电解质宏观结构设计才能找到突破和创新。通常,由于聚合物分子之间的强相互作用,未增塑的固态聚合物电解质结晶度较高,离子传输受阻,加入增塑剂可以降低聚合物链之间的引力,促进离子在其中的迁移。在Aziz等进行的一系列工作中,壳聚糖作为聚合物主体与甲基纤维素共混,选用晶格能较低的NH4I提供质子,制备出以活性炭为对称电极的双电层电容器,并证明电解质中丰富的羟基易于被甘油增塑,促进质子解离的同时为质子传导提供更多的配位位点,从而达到更好的电容性能[101-102]。
为开发出具有更高电压窗口的固态聚合物电解质,Li等[103]制备了立方相LATP粉末,并将其填充到可生物降解的聚己内酯(PCL)基质中,形成了柔性的PCL-LiClO4-LATP复合固态电解质。受益于PCL良好的电化学稳定性,CPE展现出高达5 V (vsLi/Li+) 的电压窗口。但其离子电导率即使在55 ℃下也仅有3.64×10-5S·cm-1,因此后期仍然需要借助其他手段进行改性。Zhang等[104]则以聚碳酸丙烯酯为基底,与Li6.75La3Zr1.75Ta0.25O12(LLZTO) 和LiTFSI共混后将电压窗口提升至4.6 V,同时该电解质展现出较高的离子电导率(5.2×10-4S·cm-1) 和锂离子迁移数(0.75)。以该电解质组装的LiFe0.2Mn0.8PO4/Li电池拥有相对平滑的充放电曲线、较高的库仑效率(99.81%),在80周次循环后仍有83.1%的容量保持率。
凝胶电解质由于其优良的导电性、电极-电解质界面相容性以及力学一定程度的形变能力成为柔性储能器件的潜在电解质,近年来研究人员对其的研究重点主要在于对力学性能、高低温工作性能和耐酸碱性能等的提升上。如图7所示,Ma等[105]以N,N″-亚甲基双丙烯酰胺(MBAA) 为交联剂,利用自由基聚合法制备了一种基于聚丙烯酸钠(PANa) 和纤维素的双网络水凝胶。聚合物链之间的物理缠结和共价键作用共同增强了水凝胶的力学稳定性和延伸性。而聚丙烯酸中的羟基被NaOH中和后与纤维素共同展现出良好的耐碱性。这使得水凝胶加入6 mol/L KOH和0.2 mol/L Zn(CH3COO)2形成电解质后,仍然可以拉伸到初始形态的1200%,以该电解质制备的纤维状锌空气电池在拉伸500%后仍能提供稳定的功率输出。Liu等[106]将单一化学交联(SC) 木质素水凝胶用H2SO4溶液处理后研制出具有优异伸缩性能的混合型双交联木质素水凝胶电解质。这种双交联水凝胶断裂时的压缩应力为4.74 MPa,是单交联木质素水凝胶的40倍。同时以该种水凝胶为电解质,以沉积了聚苯胺的碳布作为电极制备的柔性超级电容器表现出高的比电容和能量密度。

图7 用MBAA(交联剂)、丙烯酸酯(AA)和纤维素(增强剂)合成PANa-纤维素水凝胶电解质的工艺[105]Fig.7 Synthesis of PANa-cellulose hydrogel electrolyte from MBAA,acrylate (AA) and cellulose[105]
尽管双网络水凝胶展现出良好的力学性能和柔韧性,但是化学交联网络的共价键断裂常常造成多次伸缩后凝胶的不可逆形变[107]。以氢键缔合的琼脂作为第一网络,以疏水作用缔合的聚丙烯酰胺(PAAm) 作为第二网络,利用甲基丙烯酸硬脂酸酯(SMA) 和十二烷基磺酸钠(SDS) 的上烷基的强疏水作用使两个物理交联网络发生高度的缠绕和相互渗透,从而可以制得具有超强弹性的水凝胶,其在1000次拉伸循环后残余应变仅为6.2%,恢复2 min后残余应变降至0.4%。相比之下,传统的化学交联的琼脂/PAAm凝胶在拉伸6次后残余应变就超过了50%。利用该种凝胶电解质和聚吡咯薄膜制备的高弹性超级电容器在1000次拉伸循环后展现出良好的电容性能[108]。
凝胶电解质中的离子传输依赖于离子在溶剂中的迁移难易程度,然而对于水凝胶来说,其中的溶剂水在长期工作时往往会不可避免地从凝胶中蒸发出来,在高温下这样的脱水过程更是被极大地促进,造成电化学性能的急剧恶化;而由于水的凝固点较高,因此在低温下工作时凝胶常常发生冻结,抑制离子传输并使力学性能变差。中南大学李娟课题组合成出聚2-丙烯酰胺基-2-甲基丙烯酸(PAMPS) 和PAAm组成的双网络水凝胶,并将LiCl的乙二醇(EG) 溶液加入其中后,电解质性能在-80~120 ℃的温度范围内表现出极好的稳定性[109]。该课题组运用这种电解质制备的超级电容器在-20 ℃存放30天后电容保持率高达77.8%,而在不进行额外密封防止溶剂挥发的情况下,该电容器在80 ℃下56 h后仍然保持77.3%的容量[110]。而Mo等[107]不仅通过改进电解液降低了水凝胶的工作温度,同时还通过设计水凝胶的弹性涂层避免了高温下凝胶中水分的蒸发。他们在电解液中加入一定量的乙二醇,并在水凝胶的表面通过硅氧烷键合作用包覆了一层聚二甲基硅氧烷(PDMS) 组成的弹性涂层,如图8所示。其中乙二醇具有降低水溶液冰点的作用,而包覆的PDMS弹性涂层则利用了仿生学原理,在保证水凝胶原本的弹性和韧性的基础上起到了锁水保湿的作用,避免了水凝胶在较高温度下水分的蒸发。他们基于这种水凝胶制备的柔性Zn/MnO2电池不仅在-20 ℃下储存72 h后仍然保持90.22%以上的比容量,同时还可以在沸水中连续为电子表供电64 min,具有良好的抗冻及抗热性能。

图8 包覆PDMS弹性体涂层的水凝胶切面SEM图[107]Fig.8 SEM image of cross section of hydrogel coated with PDMS elastomer[107]
2.2 单离子导电聚合物复合体系
在双离子导电聚合物组成的电化学器件中,充放电循环的电荷转移依赖于聚合物基体中小分子电解质盐解离产生的阴阳离子方向相反的迁移,而且通常情况下阳离子的迁移速度比阴离子更慢。因此在双离子导电聚合物电解质中阳离子的迁移数往往低于0.5[111]。而且由于阴离子不参与电极反应,迁移至电极附近后会积聚形成浓度梯度和反向电场,造成器件内阻升高、循环性能下降[112]。通过限制阴离子的移动,可以使器件充放电过程中只有阳离子发生迁移,制备成单离子导电聚合物电解质(single-ion conducting polymer electrolyte,SICPE),理论上阳离子的迁移数可以接近于1[113]。以单锂离子导电聚合物为例,模拟计算证明当锂离子迁移数为1时,即使进行高倍率的充放电循环,电解质中仍然不会产生浓度极化,同时电极活性材料的利用率、功率密度、能量密度都有显著提高[114-116]。Chazalviel[117]也证明锂离子迁移数接近1时锂枝晶生长的问题也能得到有效避免。但是由于阴离子迁移受阻和锂离子解离程度低的原因,多数SICPE的离子电导率仅在10-7S·cm-1左右,因此近年来大量科研人员致力于提升SICPE的离子电导率[9]。

2.2.1 有机高分子型
有机高分子型SICPE的策略是利用共价键作用将阴离子基团固定在高分子链上,利用聚合物网络中高分子链运动能力弱的特性抑制阴离子迁移,其中提升离子电导率的关键在于引入“离子传导区”[120]。1984年,Ward等[121]分别设计了含有磺酸根阴离子和羧酸根阴离子的两种高分子锂盐,然而由于聚合物主体中缺少离子传导区,导致他们即使引入了强吸电子基团以分散羧酸根上的阴离子,离子电导率最高也只有10-6S·cm-1级别。
共混是一种简单且有效的改进方法,科研人员往往借助PEO等具有离子导电能力的物质加以改进,不仅如此,当共混加入的物质中包含Lewis碱性基团时,还可以通过与阳离子的配位作用促进聚合物盐的解离。Olmedo-Martínez等[122]证实聚三氟甲基磺酰亚胺锂(PLiMTFSI) 与PEO共混后,还可以降低PEO的结晶度,且加入低分子量的PEO结晶度下降幅度更大。因此他们利用分子量为100000的PEO和PLiMTFSI共混,使70 ℃下SICPE的离子电导率升高至2.1×10-4S·cm-1。
然而,PEO的加入极大地限制了SICPE在高压器件中的应用。Merrill等[123]通过加入导电陶瓷颗粒代替了引入PEO。他们在聚乙二醇二甲基丙烯酸酯(PEGDMA) 上接枝4-苯乙烯磺酰(三氟甲基磺酰)亚胺阴离子(STFSI-)后通过阳离子交换制备出单锂离子导电聚合物基底,拉曼光谱分析证明,加入含锂陶瓷颗粒(LiCGC),可以借助STFSI-和陶瓷颗粒的配位作用促进Li+的解离,达到增强离子电导率的目的。且随着LiCGC加入量的升高,离子电导率逐渐升高,最高可达9×10-6S·cm-1(70 ℃)。Chen等[124]在他们合成的单锂离子导电聚合物中加入LAGP颗粒,并用EC/DMC增塑,制得凝胶型SICPE。LAGP的存在赋予了Li+更多的导电通道,离子电导率提高到8.3×10-4S·cm-1,锂离子迁移数达到0.93,促进了Li+在电极上的均匀沉积,同时还提高了热稳定性和耐火性,电化学窗口达到4.6 V。
Zheng等[125]直接以有机钾盐单体聚合生成单钾离子导电聚合物,用二甲氧基乙烷(DME) 溶胀后加入Al2O3填料,在20 ℃时具有6.0×10-5S·cm-1的高离子电导率和0.87的高钾离子迁移数。Nguyen等[9]开创性地制备了一种多嵌段共聚物,其结构如图9所示。其中离子嵌段(psi-PES) 由易解离的全氟磺酰亚胺锂进行功能化改性,而高Tg嵌段(FPES) 则用以保持聚合物在宽温度范围内的机械强度。在添加EC进行增塑后,30 ℃下SICPE的离子电导率最高可达10-3S·cm-1以上。
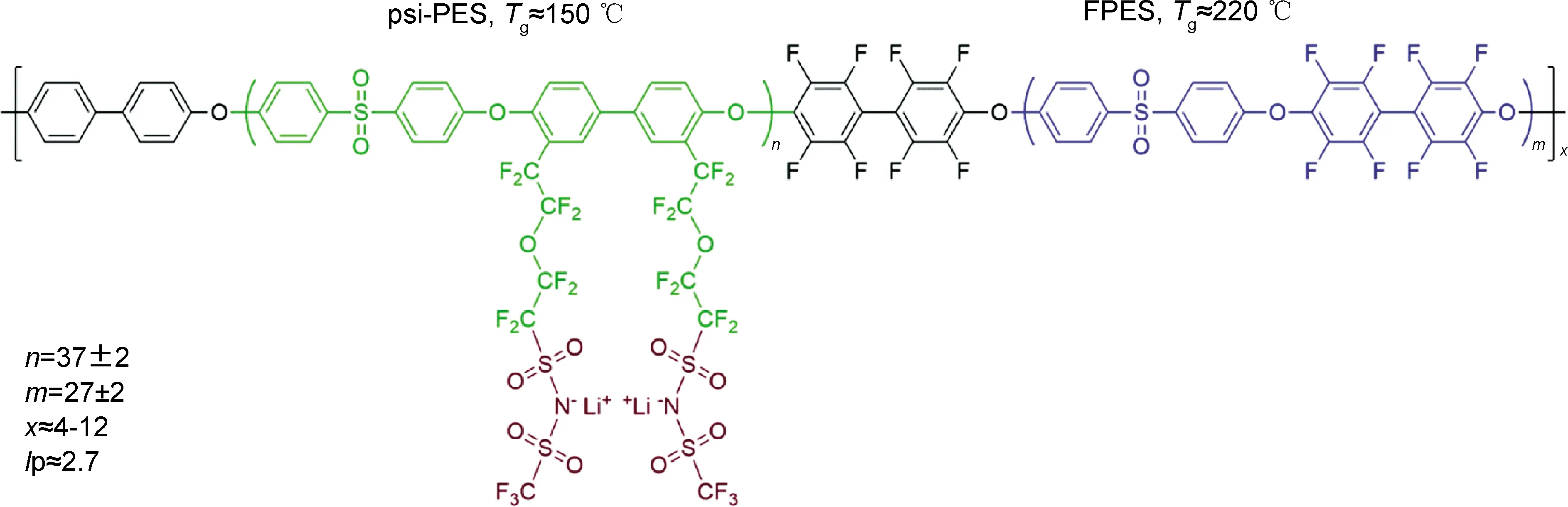
图9 单离子导电多嵌段共聚物的分子结构[9]Fig.9 Molecular structure of single ion conductive multiblock copolymers[9]
2.2.2 有机无机杂化型
在传统的固态聚合物电解质改性中,加入无机粒子以提升离子电导率和力学性能是一种常用的方法。类似的,在SICPE的改性中将阴离子接枝到无机骨架上以限制阴离子的移动,即为有机无机杂化型SICPE。
Zhao等[126]发现,当仅以聚(4-苯乙烯磺酸锂)接枝在SiO2纳米颗粒表面时,将纳米颗粒混合于PEO中制备的SICPE电导率最大仅为60 ℃下2.2×10-7S·cm-1。这是因为接枝聚合物在纳米颗粒表面形成了一层致密的网络,解离的锂离子大部分被捕捉进了网络内部,而未对整体的离子电导产生贡献。因此在SiO2表面将4-苯乙烯磺酸锂和聚乙二醇甲基丙烯酸酯(PEGMA) 共聚(如图10(a)所示)后再加入PEO中,利用PEGMA和PEO重复单元多、溶解程度大的特点,使得聚合物链在PEO基质中充分伸展,纳米颗粒表面聚合物层内部的Li+也能够参与到离子迁移中来,离子电导率提升至10-6S·cm-1数量级。
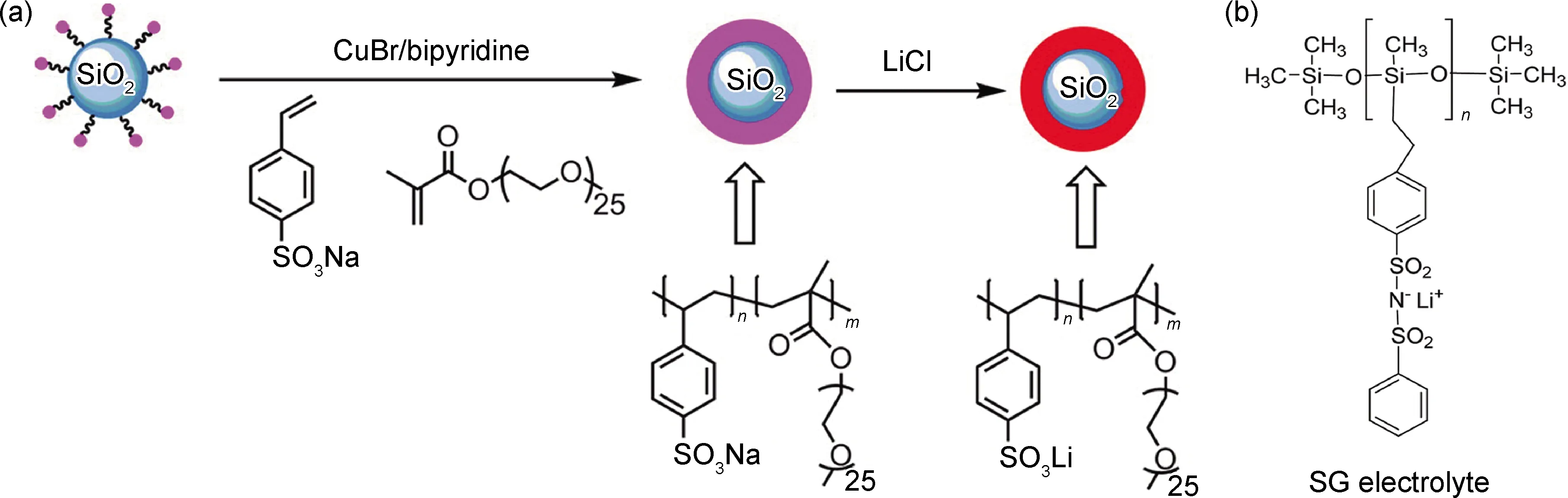
图10 有机无机杂化型SICPE设计思路(a)在SiO2颗粒表面共聚4-苯乙烯磺酸锂和PEGMA后锂化[126];(b)Rohan等合成的单离子导电聚合物[127]Fig.10 Design of organic and inorganic hybrid SICPE(a)co-polymerization of sodium 4-styrenesulfonate and PEGMA from silica nanoparticles and lithiation[126];(b)single ion conductive polymers synthesized by Rohan et al[127]
与大多数聚合物电解质和无机纳米粒子不同,聚硅氧烷本质上是一种无定形的聚合物,具有非常低的玻璃化转变温度、高的热稳定性和良好的机械强度,有助于作为骨架应用于SICPE的设计。Rohan等[127]通过硅氢化反应在聚硅氧烷上接枝了4-苯乙烯磺酰(苯磺酰基)酰亚胺,合成了如图10(b)所示的结构。这归因于磺酰亚胺基团大范围的电子离域,该电解质在锂离子迁移数高达0.89的同时离子电导率达到了7.2×10-4S·cm-1。不仅如此,其分解温度高达410 ℃,展现出极佳的热稳定性。但是仅有4.1 V的电化学窗口限制了其在高压器件中的应用。
2.2.3 阴离子受体型
1998年,Mehta等[128]发现某些带有硼原子的聚合物可以起到类似Lewis酸的作用,其中硼原子的空p轨道和阴离子中负电荷相互作用后固定了游离阴离子,使锂离子迁移数上升至0.62~0.88。这种利用相互结合的束缚作用限制阴离子运动的物质即为阴离子受体。Dai等[129]以纤维素为支撑介质,设计了具有3D结构富含硼酸盐的凝胶聚合物电解质。该电解质的锂离子迁移数为0.76,证明离子传导机理近似于单离子导电,室温离子电导率高达8.4×10-4S·cm-1。他们还证明该电解质具有抑制锂枝晶生成的作用,400周次循环后容量保持率达到89.73%,有望在新一代锂金属电池电解质中得到应用。
尽管前文已述及MOF材料在固态电解质离子电导率提高中获得了大量应用,但近年来关于改进单离子导体性能的研究仍然较少。MOF材料可以与多种阳离子兼容,具有可调节的多孔结构供给离子进行快速的迁移,是研究单离子导电的绝佳材料。Park等[130]报道了一种基于铜和偶氮唑的MOF材料,高达2066 m2/g的比表面积使其可以使多孔结构的内部结合更多的阴离子、促进阳离子的迁移。他们证明,材料对Li+,Na+,Mg2+的离子电导率分别达到4.4×10-5,1.8×10-5S·cm-1和8.8×10-7S·cm-1。这表明MOF材料具有为多种阳离子提供单离子导电的能力,未来可以与多种SICPE结合以满足不同应用场景的需求。
杯芳烃是一类亚甲基桥连苯酚单元构成的大环化合物,通过调节杯芳烃及其衍生物的结构,同样可以对不同体积的阴离子进行捕获[120]。Blazejczyk等[131]发现在LiTf/PEO体系中加入杯[4]芳烃可以使锂离子迁移数提升至0.9。但是由于杯[4]芳烃形成的空间位阻较大,因此添加量较多时将严重阻碍Li+的运输。Kalita等[132]设计了体积更小的杯[6]吡咯,他们证明将杯[6]吡咯加入掺有LiBF4的聚醚基电解质中,锂离子迁移数最大可以从0.32提升至0.92。然而杯芳烃和聚合物的相容性较差,在实际应用中为了获得较高的离子迁移数往往需要加入大量的杯芳烃,但这又导致聚合物主体的相分离,使电解质的机械强度难以满足需求,因此近年来关于杯芳烃用于SICPE的报道越来越少。
2.3 高介电常数聚合物复合体系
研究表明,高介电常数的溶剂可提高盐的离解能力和溶解度,从而提高离子电导率[133]。而聚偏氟乙烯(PVDF) 中因具有强极性的C—F键而展现出高的介电常数(ε≈8.4),同时由于其优良的力学性能、热稳定性和耐腐蚀性,广泛应用于电解质、介电材料、光伏背板等方面的研究。然而纯PVDF结晶度较高,可用于盐离子迁移的自由体积较小,室温下常常展现出低的电导率[134]。为了将其应用于固态电解质,研究人员常常通过共混、共聚等方法降低其结晶度以提升离子电导率。
清华大学南策文院士团队[135]发现将Li6.75La3Zr1.75Ta0.25O12(LLZTO) 填料引入PVDF基底后,根据图11(a)所示的拉曼光谱发现聚合物发生了脱氟化氢的反应,从而增强了PVDF基质、锂盐和LLZTO之间的相互作用。这一复合固态电解质在25 ℃下展现出5×10-4S·cm-1的高电导率,同时力学性能和热性能也得到提升。然而他们的研究也表明无机填料掺杂后PVDF的结构不稳定,受环境因素影响大,且在长期的循环过程中易发生降解,导致电化学性能下降。对此,Bag等[136]提出了在CPE中引入LiF改性的方法,改性后的CPE组装成的Li对称电池循环稳定性明显优于未改性的CPE,同时EDAX测试显示循环50 h后未改性的CPE中碳氟比C/F下降程度明显大于改性CPE,这表明LiF的引入有效防止了PVDF结构中C—F键的断裂。
研究表明,掺入陶瓷颗粒的质量分数恰好低于单独通过陶瓷颗粒进行离子传导的渗流阈值时,复合电解质的离子电导率达到最大[137]。Sun等[137]分别制备了微米尺度和纳米尺度的Nb/Al掺杂Li7La3Zr2O12(LLZO) 颗粒,并将两种颗粒以1∶1的比例混合后制备成PVDF/LLZO/LiClO4复合固态电解质。如图11(b)所示,其室温离子电导率远高于只掺杂纳米尺度或微米尺度LLZO颗粒的对照组,同时锂离子迁移数达到0.682。XPS和拉曼光谱分析表明,这来自不同尺寸LLZO颗粒与基底中PVDF和LiClO4共同作用导致的低孔隙率和长距离离子传输通道。Li等[138]在超薄PVDF膜中填充一维Li0.35La0.55TiO3(LLTO) 纳米纤维,不仅有效提高了复合膜的力学性能,同时还为离子传输提供了连续的通道,室温离子电导率提升至5.8×10-4S·cm-1。
目前受到广泛关注的体系还有聚偏氟乙烯-六氟乙烯(PVDF-HFP),该体系由结晶相的PVDF和非结晶相的HFP共聚而成,如图11(c)所示,其中结晶相的PVDF具有优良的化学稳定性和力学性能,可以保证复合电解质具有足够的机械强度;非结晶相HFP具有良好的可塑性,主要作为电解质离子传输介质。HFP的引入不仅阻碍了PVDF链段的规整排列,使结晶度大大降低,还减弱了F-的反应活性,有助于吸收更多的电解液和改善电极-电解质之间的界面稳定性[139-141]。然而即使如此,为使该体系在电解质中具有实际应用价值,仍需要进行大量的改性研究。
最早进行产业化应用的技术由Bellcore公司开发,使用PVDF-HFP制成多孔薄膜,吸收大量电解液后展现出良好的离子电导率和力学性能。但是由于PVDF-HFP在较高的温度条件下会发生溶解,因此近年来研究人员开发了一系列的方法增强其热稳定性。Xiao等[142]在PVDF-HFP体系中共混添加了聚苯乙烯-聚氧乙烯-聚苯乙烯三嵌段共聚物(PS25-PEO-PS25),利用Friedel-Crafts烷基化反应进行化学交联,制备出超交联多孔聚合物膜(CPMs)。PEO链段的引入促进了Li+的迁移,提高了CPMs的孔隙率和电解液吸收率,室温离子电导率最高可达2.11 mS·cm-1,并且在300 ℃下仍然保持了30%的孔隙率,这有助于其在高温环境下的进一步应用。
PVDF-HFP/聚苯胺(PANI) 共混膜表现出高的离子电导率(1.96 mS·cm-1)、孔隙率(83%) 和电解液吸收率(270%)[14]。但是PANI的加入降低了膜的力学性能,因此Ahmad等[143]在该体系中加入氧化石墨烯(GO) 纳米粒子进行改进,由此制成了PVDF-HFP/PANI/GO三元共混膜,尽管损失了部分离子电导率(下降至0.664 mS·cm-1),但拉伸强度从2.8 MPa提升至8.8 MPa。
Wang等[144]将PVDF-HFP与TPU,PMMA共混后加入Na3Zr2Si2PO12填料,制备出凝胶聚合物电解质用于钠离子电池。其中。填料的加入降低了聚合物基体的结晶度,提高了凝胶中的孔隙率,使得电解液的吸收率更大。测试表明,该凝胶电解质有高达2.83×10-3S·cm-1的离子电导率和5.16 V的电化学窗口。由其组装的钠离子0.5 C下循环100周次后容量保持率为99.2%,展现出优异的循环性能和倍率性能。为了提高无机粒子在聚合物中的分散性,Khan等[145]首先用PMMA对SiO2纳米粒子进行改性,然后加入PVDF-HFP中制备成凝胶电解质。受益于PMMA和PVDF-HFP之间良好的相容性,SiO2颗粒在基体中均匀分散,当掺杂量为10%时,离子电导率达到2.22×10-3S·cm-1。
区别于聚合物的简单共混体系,Luo等[141]报道了一种由线性PVDF-HFP和紫外交联季戊四醇四丙烯酸酯(PETEA) 组成的半互穿聚合物凝胶网络,如图11(d)所示,PETEA单体中具有四个交叉的C—C键,在凝胶聚合物中形成交联网络结构,为离子传输提供了更多的通道,从而展现出较高的离子电导率(0.5 mS·cm-1) 和较宽的电化学窗口(4.8 V)。Pan等[146]用离子液体1-乙基-3-甲基咪唑双三氟甲磺酰亚胺盐(EMIMTFSI)掺杂制备了凝胶聚合物电解质EMIMTFSI/PVDF-HFP,并组装成纤维状非对称超级电容器。凝胶聚合物电解质极佳的力学性能使器件在90°弯曲1000个循环后仍然有92.7%的容量保持率,并且离子液体的引入使器件的电化学窗口提升至3.5 V,为可穿戴电源器件的开发提供了新的途径。
3 结束语
本文从聚合物研究改性的角度出发,综述了不同种类聚合物在电化学器件电解质中的应用,以及近年来对这些聚合物电解质的改性策略,以期有助于未来相关领域的研究。要想克服当前液态电解质普遍表现出来的安全性差、环境危害大等缺陷,并适应新一代电化学器件对高柔韧性、高能量密度、高安全性等要求,电解质由液态转向固态是必然的趋势。为了顺应这一趋势,有必要设计出具有安全保证和独特应用效益的新型SPE。但是当前的研究中,仍然存在着较多的问题限制了SPE的进一步商业应用,本文将这些挑战梳理如下,以此为今后的研究提供进一步破解的信息,并提出了解决这些问题的一些初步设想。
(1)理论研究的匮乏。当前尽管已经存在大量的模型对SPE中的导电机理进行了阐释,但是这些阐释一方面不够具有普遍性,很多计算过程仍然需要依赖引入经验常数,当聚合物基体或其他因素发生变化时,这些模型往往不能准确反映其导电机理的变化情况;另一方面这些模型主要围绕本征聚合物电解质展开,对于共混、共聚等改性手段对SPE性能的影响机理并没有充分的研究。
(2)离子电导率相对较低。离子电导率低的问题极大地限制着固态电化学器件的充放电能力。目前,有机液态电解质的离子电导率可以达到10-2~10-3S·cm-1,SPE要想进入商业化应用也需要将电导率尽可能提升到10-4S·cm-1以上。尽管凝胶聚合物电解质可以比较轻易地将电导率提升至这一水平,但只能作为电解质由液态转向固态的过渡产品使用,其本身所面临的高低温性能差、力学性能差等缺点仍然需要克服。
(3)阳离子迁移数有待提升。高的阳离子迁移数有助于降低器件内部的极化程度、抑制枝晶的生长,但同时阳离子迁移数的提升必然伴随阴离子迁移受阻,这将使得SPE的离子电导率进一步下降。如何同时兼顾高阳离子迁移数和高离子电导率这一对矛盾,仍需研究人员进行进一步的开发与改性。
(4)热稳定性和化学稳定性的不足。新一代电化学器件需要具有在各种极端环境下工作的能力,如极端高低温、强酸、强碱等,提升电解质的热稳定性和电化学稳定性将提高器件的环境适应性。
(5)生物兼容性的不足。随着可穿戴电子设备和体内移植电子设备的应用,未来需要寻找具有更好的生物相容性,且能满足电解质基本要求的聚合物,从而提高这些生物医学装置的稳定性和可靠性。
因此,在未来的研究中需要综合考虑这些问题,以期分步或部分解决这些挑战。如对于理论欠缺问题,可以考虑通过引入先进表征测试技术获得大量实验数据,通过实验数据的拟合建立经验或半经验方程,或以大量实验数据作为边界条件,结合密度泛函理论计算、分子动力学模拟等技术,建立模拟方程,由此对SPE及其复合体系中尚未发现的理论问题进行进一步的探索;而对于离子电导率相对较低、阳离子迁移数以及热或化学稳定性等问题,可以考虑通过合成新型离子导体并将其与聚合物基体复合构筑特殊的微纳米多相体系、或在已有SPE体系中采用多种离子注入技术以及通过超分子组装方式构筑特殊的分子复合体系提高其离子电导率,平衡阳离子迁移数与离子电导率之间矛盾,且同时提升热学及化学稳定性等;而对于可穿戴领域中的聚合物固态电解质,则需要针对已有生物高分子体系中不足,采用新型的生物高分子基体或通过分子杂化或复合方法构筑生物相容性复合材料体系。
综上所述,注重对具有特殊结构的新型聚合物或功能助剂的设计,并以此构筑具有独特的分子杂化及微纳米复合结构,有望满足SPE电化学性能要求同时赋予体系其他所需性能,如耐高压性能、力学性能、抗高低温性能等,实现SPE更高的应用价值。
