提高感冒疏风丸质量标准的研究
2022-05-07杨深应刘常逊王洪云陶明宝曹朴琼丁国瑜赵兴蕊
杨深应,刘常逊,王洪云,陶明宝,曹朴琼,丁国瑜,杨 麒,赵兴蕊*
1.保山市食品药品检验检测中心,保山 678000;2.保山中医药高等专科学校,保山 678000
感冒疏风丸由麻黄绒(炙)、苦杏仁、桂枝、白芍(酒炙)、紫苏叶等12味药材制成,具有辛温解表、宣肺和中的功效,临床常用于治疗风寒感冒、发热咳嗽、头痛怕冷、鼻流清涕等症[1]。处方中君药为麻黄绒(炙),其主要有效成分为盐酸麻黄碱和盐酸伪麻黄碱,具有发汗、镇咳、平喘、强心、升高血压、抗炎、抗病毒等药理作用,临床用于感冒、支气管哮喘、某些低血压状态、皮肤黏膜疾病等的治疗[2]。目前感冒疏风丸仅有昆明中药厂有限公司生产,有水蜜丸和大蜜丸2种剂型。
感冒疏风丸的现行质量标准收载于《卫生部药品标准中药成方制剂》第十三册,其标准项下仅有性状、显微、芍药苷的薄层色谱(thin layer chromatography,TLC)鉴别及检查项目,无含量测定项目[1],目前感冒疏风丸已成为以风寒表证为主的新型冠状病毒肺炎早期干预的中成药之一[3]。动物实验结果显示,由相同药味组成的不同剂型的感冒疏风片对金黄色葡萄球菌、大肠杆菌、肺炎球菌等具有抑菌作用,且具有抗炎、镇痛、解热的作用[4],但有关感冒疏风丸质量控制方面的研究报道较少[5],现有标准已不适用于现代中药质量评价与控制的需求,为确保感冒疏风丸的质量可控、疗效可靠,急需提高其质量标准。本实验通过对感冒疏风丸的质量标准进行系统研究,建立了麻黄绒(炙)、桂枝、紫苏叶、甘草、独活的TLC鉴别方法及同时测定盐酸麻黄碱和盐酸伪麻黄碱含量的HPLC法。
1 仪器与试药
1.1 仪器
Agilent 1260型高效液相色谱仪(配DAD 检测器,美国安捷伦公司);CP225D 型电子天平(德国赛多利斯公司);DZKW-S-4型电热恒温水浴锅(北京市永光明医疗仪器有限公司);SK2510LHC 型超声波清洗器(上海科导超声仪器有限公司);BPG-9140A型鼓风干燥箱(上海一恒科学仪器有限公司);Good-Look-2000型薄层色谱成像系统(上海科哲生化科技有限公司);硅胶G 薄层板(批号20200323,烟台市化学工业研究所)。
1.2 试药
13批感冒疏风丸(水蜜丸规格为6 g/袋,批号分别 为200717、280780、281810、280262、271362、271285;大蜜丸规格为9 g/丸,批号分别为100070、180138、190024、190089、180136、190116、180187,昆明中药厂有限公司)。以下对照药材和对照品均购自中国食品药品检定研究院:对照药材麻黄(草麻黄,批号121051-201606);麻黄(中麻黄,批号121604-201502);桂枝(批号121191-201906);甘草(批号120904-202021);独活(批号120940-201612);对照品盐酸麻黄碱(批号171241-201508,质量分数为99.8%);对照品盐酸伪麻黄碱(批号171237-201510,质量分数为99.8%);对照品桂皮醛(批号110710-201821);对照品二氢欧山芹醇当归酸酯(批号111583-201605)。甲醇、乙腈为色谱纯,均购自默克股份两合公司;其他试剂为分析纯;水为超纯水。
2 方法与结果
2.1 麻黄绒(炙)TLC鉴别
2.1.1 制备供试品溶液 取研碎的水蜜丸6 g,或取剪碎的大蜜丸9 g,加5 g硅藻土研匀[6];加适量氨水使其润湿,再加40 mL二氯甲烷回流1 h,过滤,蒸干滤液,残渣加1 mL甲醇溶解,即得[7]。
2.1.2 制备阴性样品溶液 取按感冒疏风丸处方比例及制法制备的缺麻黄绒(炙)的阴性样品9 g,按2.1.1项下方法制备溶液。
2.1.3 制备对照药材溶液 取麻黄(草麻黄)、麻黄(中麻黄)对照药材各1 g,按照2.1.1项下方法制备溶液。
2.1.4 制备对照品溶液 取盐酸麻黄碱对照品适量,加甲醇制成质量浓度为1 mg·mL-1的溶液。
2.1.5 鉴别方法[8]吸取上述4 种溶液各5~10μL,分别点于同一硅胶G 薄层板上,展开剂为氯仿-甲醇-甲酸(16∶4∶4),展开约8 cm,喷以体积分数为0.5%的茚三酮乙醇溶液,于105℃加热至斑点显色清晰。结果见图1。在与麻黄对照药材、盐酸麻黄碱对照品色谱对应的位置上,13批样品均出现相同的紫红色斑点,且阴性无干扰。

图1 麻黄绒(炙)鉴别的TLC图Fig.1 TLC chromatograms of Ephedrae Herba Velvet(roast)identification
2.2 桂枝TLC鉴别[7-9]
2.2.1 制备供试品溶液 取研碎的水蜜丸18 g,或取剪碎的大蜜丸27 g;加50 mL甲醇超声40 min,过滤,滤液置于50℃水浴中蒸干,残渣加30 mL 水溶解,用乙醚振摇提取4次,每次20 mL,合并乙醚液,挥干(水液备用),残渣加2 mL甲醇溶解,即得[9]。
2.2.2 制备阴性样品溶液 取按感冒疏风丸处方比例及制法制备的缺桂枝阴性样品27 g,按照2.2.1项下方法制备溶液。
2.2.3 制备对照药材溶液 取桂枝对照药材1 g,按照2.2.1项下方法制备溶液。
2.2.4 制备对照品溶液 取桂皮醛对照品适量,加甲醇制成质量浓度为1 mg·mL-1的溶液。
2.2.5 鉴别方法[8]吸取上述4种溶液各10~15μL,分别点于同一硅胶G 薄层板上,展开剂为石油醚(60~90℃)-乙酸乙酯(17∶3),展开约8 cm,喷以二硝基苯肼乙醇试液[7]。结果见图2。在与桂枝对照药材、桂皮醛对照品色谱对应的位置上,13批样品均出现相同的橙色至橙红色斑点,且阴性无干扰。

图2 桂枝鉴别的TLC图Fig.2 TLC chromatograms of Cinnamomi amulus identification
2.3 紫苏叶TLC鉴别
2.3.1 制备供试品溶液 取研碎的水蜜丸12 g,或取剪碎的大蜜丸18 g;加20 mL水研匀,移入500 mL圆底烧瓶中,加水230 mL,依照挥发油测定法实验,加入1.5 mL石油醚(60~90℃),加热并保持微沸2 h,取石油醚层作为供试品溶液[7]。
2.3.2 制备阴性样品溶液 取按感冒疏风丸处方比例及制法制备的缺紫苏叶阴性样品18 g,按照2.3.1项下方法制备溶液。
2.3.3 制备对照药材溶液 取紫苏叶对照药材1 g,按照2.3.1项下方法制备溶液。
2.3.4 鉴别方法[8]吸取上述3种溶液各10μL,分别点于同一硅胶G 薄层板上,展开剂为石油醚(60~90℃)-乙酸乙酯(30∶2),展开约8 cm,喷以体积分数为5%的香草醛盐酸溶液,于105℃加热至斑点显色清晰。结果见图3。在与紫苏叶对照药材色谱对应的位置上,13批样品均出现相同颜色的斑点,且阴性无干扰。

图3 紫苏叶鉴别的TLC图Fig.3 TLC chromatograms of Perillae Folium identification
2.4 独活TLC鉴别
2.4.1 制备供试品溶液 取研碎的水蜜丸6 g,或取剪碎的大蜜丸9 g,加5 g硅藻土研匀;加50 mL水超声30 min,再加入50 mL 石油醚(60~90℃)超声30 min,分取石油醚液蒸干,残渣加1 mL 甲醇溶解,即得[5]。
2.4.2 制备阴性样品溶液 取按照感冒疏风丸处方比例及制法制备的缺独活阴性样品9 g,按照2.4.1项下方法制备溶液。
2.4.3 制备对照药材溶液 取独活对照药材1 g,按照2.4.1项下方法制备溶液。
2.4.4 制备对照品溶液 取二氢欧山芹醇当归酸酯对照品适量,加甲醇制成质量浓度为0.5 mg·mL-1的溶液。
2.4.5 鉴别方法[8]吸取上述4种溶液各5~10μL,分别点于同一硅胶G薄层板上,展开剂为石油醚(60~90℃)-乙酸乙酯(20∶10),展开约8 cm,置于365 nm紫外光下检视。结果见图4。在与独活对照药材、二氢欧山芹醇当归酸酯对照品色谱对应的位置上,13批样品均出现相同的亮蓝色荧光斑点,且阴性无干扰。
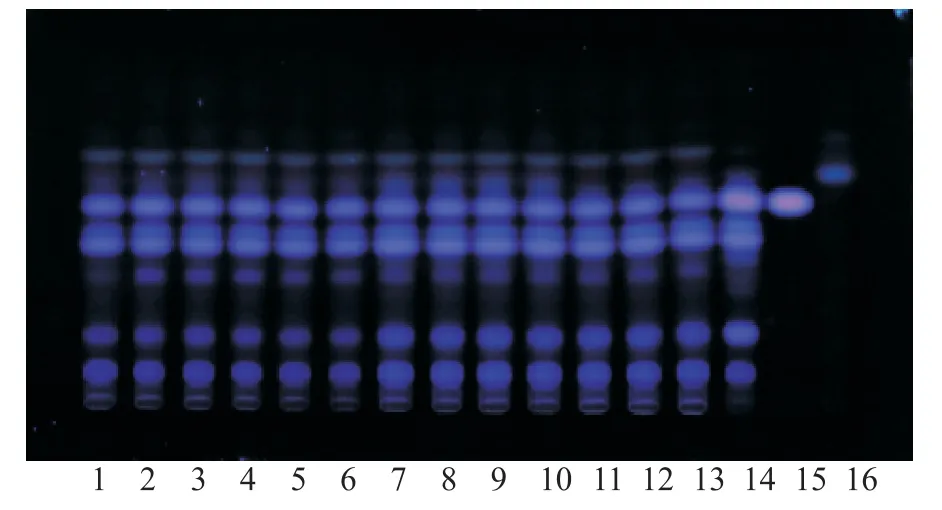
图4 独活鉴别的TLC图Fig.4 TLC chromatograms of Angelicae ubescentis Radix identification
2.5 甘草TLC鉴别
2.5.1 制备供试品溶液 取2.1.2项下桂枝的水液[9],用水饱和的正丁醇振摇提取4次,每次20 mL,合并正丁醇液,用水洗涤3次,每次20 mL,蒸干正丁醇液,残渣加2 mL甲醇溶解,即得[6]。
2.5.2 制备阴性样品溶液 取按照感冒疏风丸处方比例及制法制备的缺甘草阴性样品27 g,按照2.5.1项下方法制备溶液。
2.5.3 制备对照药材溶液 取甘草对照药材1 g[7],按照2.5.1项下方法制备溶液。
2.5.4 鉴别方法[8]吸取上述3 种溶液各2~5μL,分别点于同一硅胶G 薄层板上,展开剂为乙酸乙酯-冰醋酸-甲酸-水(30∶2∶2∶4),展开约8 cm,喷以体积分数为10%的硫酸乙醇溶液,于105℃加热至斑点显色清晰,分别置于日光下和365 nm 紫外光下检视。结果见图5。在与甘草对照药材色谱对应的位置上,13批样品均出现相同颜色的主斑点或荧光主斑点,且阴性无干扰。

图5 甘草鉴别的TLC图Fig.5 TLC chromatograms of Glycyrrhizae Radix Et Rhizoma identification
2.6 含量测定
2.6.1 色谱条件 色谱柱:Agilent Eclipse Plus Phenyl-Hexyl(250 mm×4.6 mm,5μm);柱温:30℃;检测波长:207 nm;流动相:乙腈-1 mL·L-1磷酸溶液(2∶98);流速:1.0 mL·min-1;进样量:10μL。
2.6.2 混合对照品溶液的制备 精密称取对照品盐酸麻黄碱22.03 mg、盐酸伪麻黄碱21.24 mg,分别置于50 mL 量瓶中,加甲醇溶解、稀释至刻度,得单一对照品母液。精密量取盐酸麻黄碱对照品母液1 mL、盐酸伪麻黄碱对照品母液0.5 mL,置于同一20 mL 量瓶中,加含体积分数1.44%磷酸的体积分数70%甲醇溶液稀释至刻度,得盐酸麻黄碱、盐酸伪麻黄碱质量浓度分别为21.99、10.60μg·mL-1的混合对照品溶液。
2.6.3 供试品溶液的制备 取水蜜丸,研细,取约1.5 g,精密称定,或取质量差异项下的大蜜丸,剪碎,取约2 g,精密称定。精密加入25 mL 含体积分数1.44%磷酸的体积分数70%甲醇溶液,称质量,放置过夜,超声处理(功率250 W,频率35 k Hz)60 min使药物全部溶散,放冷,再称质量,用含体积分数1.44%磷酸的体积分数70%甲醇溶液补足减失的质量,摇匀,过滤,取续滤液,即得。
2.6.4 阴性样品溶液的制备 取按照感冒疏风丸处方比例及制法制备缺麻黄绒(炙)的阴性样品,按照2.6.3项下方法制得缺麻黄绒(炙)的阴性样品溶液。
2.6.5 专属性考察 精密吸取上述3 种溶液各10μL,注入液相色谱仪,按照2.6.1项下色谱条件分析,结果见图6。理论塔板数按盐酸麻黄碱或盐酸伪麻黄碱峰计算均不低于5 000,各色谱峰分离良好,阴性对照无干扰。

图6 含量测定的HPLC图Fig.6 HPLC chromatograms of content determination
2.6.6 线性关系考察 取盐酸麻黄碱、盐酸伪麻黄碱质量浓度分别为87.94、42.40μg·mL-1的混合对照品溶液1、2、5、8、10、15、20 mL,分别置于20 mL量瓶中,加含体积分数1.44%磷酸的体积分数70%甲醇溶液定容,按照2.6.1项下色谱条件进样测定。以溶液质量浓度(μg·mL-1)为横坐标(x)、峰面积为纵坐标(y)进行回归,得盐酸麻黄碱、盐酸伪麻黄碱的回归方程分别为y1=2 420.5x1+3.846 2(r=0.999 9)和y2=2 449.5x2+4.025 6(r=0.999 92),盐酸麻黄碱、盐酸伪麻黄碱质量浓度分别在4.40~87.94、2.12~42.40μg·mL-1范围内呈良好的线性关系。
2.6.7 精密度实验 取2.6.2项下混合对照品溶液,按照2.2.1项下色谱条件重复测定6次。计算得盐酸麻黄碱、盐酸伪麻黄碱峰面积的RSD 值分别为0.44%、0.64%,表明仪器的精密度良好。
2.6.8 重复性实验 取水蜜丸(批号200717),按照2.6.3项下方法制备供试品溶液,平行6份,按照2.6.1项下色谱条件进样测定。计算得盐酸麻黄碱、盐酸伪麻黄碱含量的平均值分别为0.348 4、0.193 4 mg·g-1,RSD值分别为0.91%、0.98%,表明该方法的重复性良好。
2.6.9 稳定性实验 取水蜜丸(批号200717),按照2.6.3项下方法制得供试品溶液,于0、4、8、12、18、24 h按照2.6.1项下色谱条件进样测定。计算得盐酸麻黄碱、盐酸伪麻黄碱峰面积的RSD 值分别为1.27%、1.65%,表明溶液在24 h内稳定。
2.6.10 加样回收率实验 取已知含量的水蜜丸(批号200717)适量,研细,精密称取0.75 g,共6份,精密加入盐酸麻黄碱、盐酸伪麻黄碱质量浓度分别为0.052 8、0.029 7 mg·mL-1的混合对照品溶液5 mL和含体积分数1.44%磷酸的体积分数70%甲醇溶液20 mL,按照2.6.3项下方法制备供试品溶液,按照2.6.1项下色谱条件进样测定,计算回收率。结果见表1。

表1 回收率实验结果 (n=6)Tab.1 Results of recovery tests(n=6)
2.6.11 样品含量测定 分别取6批水蜜丸及7批大蜜丸,每批3份,按照2.6.3项下方法制备供试品溶液,按照2.6.1项下色谱条件进样测定,计算盐酸麻黄碱、盐酸伪麻黄碱的含量及总量。结果见表2。
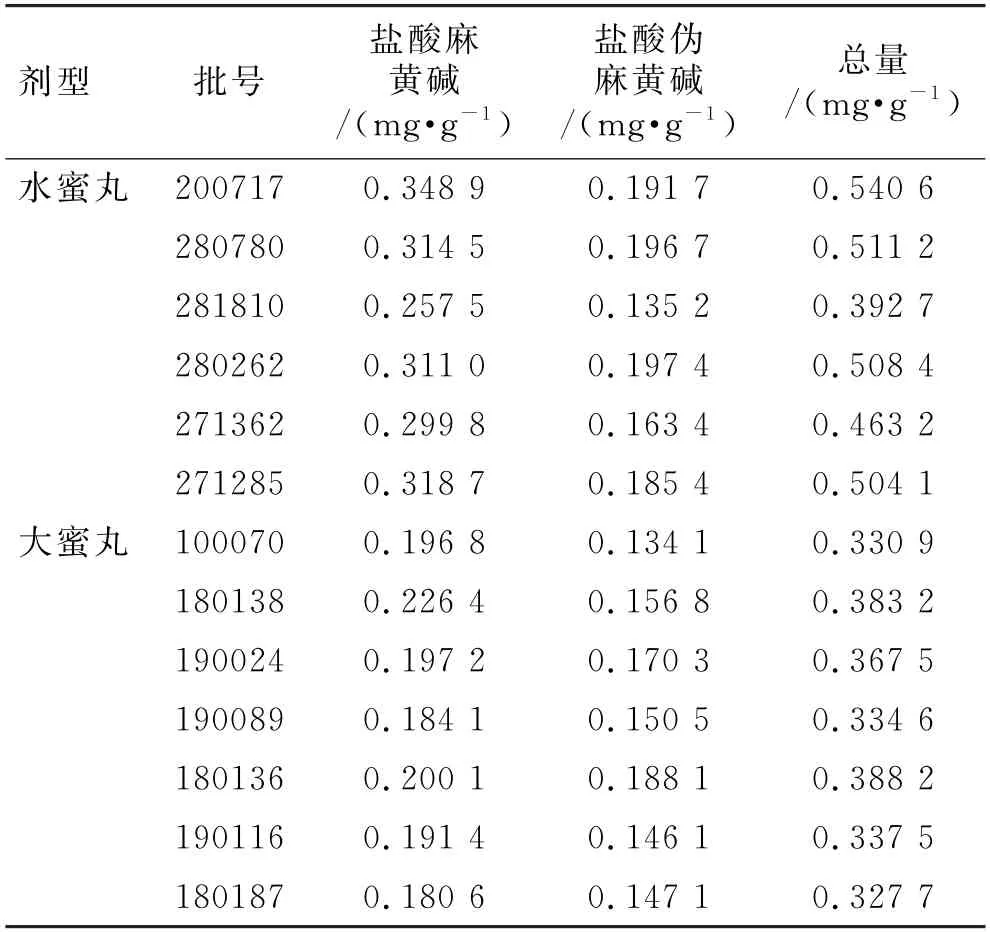
表2 样品测定结果 (n=3)Tab.2 Results of sample determination(n=3)
3 讨论
3.1 薄层色谱鉴别方法的考察
感冒疏风丸方中药味较多,成分复杂,在进行桂枝、紫苏叶TLC鉴别时,参照《中华人民共和国药典》(以下简称《中国药典》)2020年版一部桂枝、紫苏叶项下TLC鉴别方法,结果阴性对照干扰较大。因此,在桂枝TLC鉴别实验中,分别对乙醇、甲醇、乙酸乙酯、乙醚、石油醚(60~90℃)、石油醚(30~60℃)等提取溶剂和振摇、超声、加热回流、挥发油提取等提取方法的前处理条件进行摸索,最终选择甲醇提取,蒸干后水溶解,再用乙醚萃取的提取方法,该方法能够排除阴性对照的干扰;在紫苏叶TLC鉴别实验中,对正己烷-乙酸乙酯、乙酸乙酯-甲醇-甲酸-水、环己烷-乙酸乙酯、石油醚(60~90℃)-乙酸乙酯等不同展开系统及二硝基苯肼乙醇溶液、体积分数10%硫酸乙醇溶液、体积分数1%香草醛硫酸溶液、体积分数5%香草醛盐酸溶液等显色剂进行摸索,结果显示以石油醚(60~90℃)-乙酸乙酯(15∶1)为展开剂,以体积分数5%香草醛盐酸溶液为显色剂时,才能排除阴性对照的干扰。
3.2 含量测定方法的考察
3.2.1 提取条件的选择 查阅相关文献[7-15],本实验分别比较了以下提取条件对提取结果的影响:不同提取溶剂(含体积分数1.44%磷酸水溶液、含体积分数1.44%磷酸的体积分数70%甲醇溶液、含体积分数1.44%磷酸的体积分数50%甲醇溶液、含体积分数1.44%磷酸的甲醇溶液等),不同提取方法(超声、回流、超声后回流、放置过夜后超声等)提取。最终确定以含体积分数1.44%磷酸的体积分数70%的甲醇溶液为提取溶剂,放置过夜后超声60 min的提取方法。
3.2.2 色谱条件的选择 在查阅相关文献的基础上[7-18],考察了甲醇-体积分数0.092%磷酸溶液(含体积分数0.04%三乙胺和体积分数0.02%二正丁胺)、乙腈-体积分数0.3%三乙胺的0.02 mol·L-1磷酸二氢钾溶液、乙腈-体积分数0.2%磷酸溶液-三乙胺、乙腈-体积分数0.1%磷酸溶液等不同比例的流动相,结果显示,以乙腈-体积分数0.1%磷酸溶液(2∶98)为流动相时,待测成分的出峰时间、峰形及分离度等较好。同时对以下色谱柱进行了考察:以十八烷基硅烷键合硅胶为填充剂的色谱柱Diamonsil Plus C18(250 mm×4.6 mm,5μm)、SHISEIDO AQ C18(250 mm×4.6 mm,5μm)、Agilent ZORBAX SB-Aq C18(250 mm×4.6 mm,5μm);以极性乙醚连接苯基键合硅胶为填充剂的色谱柱Agilent Eclipse Plus Phenyl-Hexyl(250 mm×4.6 mm,5μm)、FeiniGen Ruby Phenyl(250 mm×4.6 mm,5μm)。结果显示,使用极性乙醚连接苯基键合硅胶柱获得的峰形较好,故选择Agilent Eclipse Plus Phenyl-Hexyl(250 mm×4.6 mm,5μm)色谱柱进行后续实验。
3.3 含量限度的拟定
麻黄绒(炙)的主要有效成分为盐酸麻黄碱与盐酸伪麻黄碱,二者为同分异构体,在麻黄中所占的比例不呈恒定状态,参考《中国药典》2020年版一部麻黄含量测定项目[7]、感冒疏风丸的处方及制法[1],结合本实验含量测定的结果,暂拟定含量限度为感冒疏风丸含麻黄绒(炙)以盐酸麻黄碱和盐酸伪麻黄碱的总量计,水蜜丸每1 g不得少于0.31 mg;大蜜丸每1 g不得少于0.20 mg。
综上所述,本研究在感冒疏风丸原质量标准的基础上,增加了麻黄绒(炙)、桂枝、紫苏叶、甘草、独活的TLC鉴别方法,用HPLC法测定了盐酸麻黄碱和盐酸伪麻黄碱的含量,提高了感冒疏风丸的质量控制标准。
