大鼠脑缺血再灌注通过TGF- β1/Smad7通路诱发急性肾损伤的机制探究
2022-04-25刘云方锦程钟文华王林张艺洪云李曙
刘云,方锦程,钟文华,王林,张艺,洪云,李曙
(1.皖南医学院 病理生理学教研室,安徽 芜湖 241001; 2.皖南医学院弋矶山医院 神经外科,安徽 芜湖 241001; 3.皖南医学院弋矶山医院 超声医学科,安徽 芜湖 241001)
急性脑卒中是临床上较为常见的脑血管疾病,它可以发生在机体器官或组织缺血缺氧过程及缺血再灌注(ischemia reperfusion,IR) 治疗后[1]。大量研究证明,脑IR可活化炎症细胞,促进炎症介质释放至远端器官,最终引起全身性炎症反应[2- 3],其中肾脏损伤也较为多见。目前已有研究证明导致脑组织IR损伤的机制有白细胞聚集、自由基生成和细胞内超载等[4- 5],但对肾脏组织损伤的机制仍缺乏明确的研究结果,导致临床在进行治疗和药物干预时缺乏理论支持,所以脑IR引起的急性肾损伤(AKI)仍然是临床医生面临的一个严峻挑战。造成这种临床现状的一个主要原因是缺乏有效的动物模型,因此本研究希望建立一个稳定、有效、符合临床的脑IR损伤诱发AKI的动物模型,探索其发生机制,为临床治疗这一类急重并发症提供理论依据。
1 材料与方法
1.1 实验动物与试剂
选取4~6周龄SPF级健康雄性大鼠,体重240~270 g,由长沙生物天勤有限公司提供,在皖南医学院实验动物中心饲养。动物实验经安徽省动物伦理委员会批准。尿素氮(BUN)和血肌酐(Scr)检测试剂盒购自南京建成生物工程研究所。转化生长因子- β1(TGF- β1)、Smad7 抗体购自博奥森(Bioss)生物技术有限公司(北京)。Q- PCR试剂盒购于诺唯赞(Vazyme)生物科技有限公司(南京)。
1.2 实验方法
1.2.1 建立大鼠脑缺血模型 大鼠腹腔注射10%水合氯醛(0.3 ml·100 g-1)麻醉,仰卧位固定后颈部备皮消毒。沿颈正中部切口,钝性分离至暴露颈总动脉(CCA)、颈外动脉(ECA)和颈内动脉(ICA),将血管与周围的组织钝性分离,用微血管夹夹闭CCA的近心端。在ECA近交叉部位用丝线打一活结,远心端打结,之间剪开缺口插入线拴,固定活结,调整方向插至大脑中动脉分叉处,松除微血管动脉夹,此时大鼠大脑中动脉处于闭塞状态。将20只大鼠随机分为正常组和缺血2、4、6 h组4组各5只。闭塞时间达到实验所需时间时观察各组大鼠行为学表现,当大鼠出现跛行、转圈或偏瘫即为模型构建成功。此时将大鼠处死,通过肾功能及病理学观察选出损伤较为严重的时间点作为IR模型中的缺血时间点。之后取25只大鼠构建该缺血模型,并随机分为再灌注3、6、12、24、48 h 5组。根据分组时间点取出栓子并处死大鼠。
1.2.2 生化指标及肾脏质量指数检测 大鼠麻醉后腹主动脉取血,常温下以3 000 r·min-1离心10 min后获取血清。检测血清BUN和Scr水平。大鼠处死后剥离左侧肾脏称取质量,肾脏质量与处死前大鼠体重之比即为大鼠的肾脏质量指数。
1.2.3 肾脏病理学观察 大鼠处死后取出脑组织和肾组织,冲洗后用4%多聚甲醛浸泡固定48 h。依次脱水、石蜡包埋,常规HE染色和Masson染色。HE染色后镜下观察肾脏病理改变,Masson染色评估肾脏组织胶原沉积情况。
1.2.4 ELISA法检测大鼠血浆炎症因子肿瘤坏死因子- α(TNF- α)和白细胞介素6(IL- 6)水平变化 按照ELISA试剂盒说明书提取大鼠血浆,根据说明书准备标准品、样本、抗体和酶试剂,加入样本和抗体等温育后洗涤,再依次加入显色液和终止液,并立即在酶标仪波长450 nm和630 nm处检测吸光度(OD)值,用标准品的浓度和OD计算出标准曲线方程,再计算样本浓度。
1.2.5 定量PCR检测mRNA表达水平 取肾脏组织皮质区加入1 ml Trizol充分裂解,依次加氯仿、异丙醇、75%乙醇提取RNA,逆转录后得到cDNA。反应体系为:SYBR 10 μl,无RNA酶水7.2 μl,引物上、下游各 0.4 μl,cDNA 2 μl。反应条件:95 ℃ 3 min,95 ℃ 20 s,60 ℃ 20 s,共40个循环。β- 肌动蛋白(β- actin)为对应内参,采用 2-ΔΔCt计算出目的基因相对表达量。引物序列如下:TGF- β1正向5′- TGCGCTTGCAGAGATTAAAA- 3′,反向5′- AGCCCTGTATTCCGTCTCCT- 3′;Smad7正向5′- CAGATTCCCAACTTCTTCTG- 3′,反向5′- GTTGAAGATGACCTCCAGCC- 3′;胶原蛋白Ⅰ正向5′- AGCACGTCTGGTTTGGAGAG- 3′,反向5′- GACATTAGGCGCAGGAAGGT- 3′;胶原蛋白Ⅲ正向5′- ACGTAAGCACTGGTGGACAG- 3′,反向5′- CAGGAGGGCCATAGCTGAAC- 3′;β- actin正向5′- GCGCAAGTACTCTGTGTGGA- 3′,反向5′- AGGGTGTAAAACGCAGCTCAG- 3′。
1.2.6 蛋白质印迹实验 常规提取肾脏组织皮质区总蛋白,BCA法测定蛋白浓度;同体积的样本加入十二烷基硫酸钠- 聚丙烯酰胺凝胶中电泳分离蛋白后,半干法转膜;5%脱脂奶粉封闭1.5 h后加入一抗(1∶1 000) 4 ℃孵育过夜,二抗(1∶10 000)室温孵育1 h后用ECL法显色。
1.3 统计学处理
2 结 果
2.1 不同缺血时间血清BUN和Scr水平比较及肾脏组织病理学变化
血清BUN和Scr水平正常组在正常范围内,缺血4 h和6 h组较正常组和缺血2 h组显著升高(P<0.05),缺血4 h组与缺血6 h组相比差异无统计学意义,见表1;缺血4 h和6 h时肾脏质量指数较其他组显著下降。上述结果提示,肾脏功能在缺血4 h时开始出现明显改变。
表1 4组大鼠血清BUN和Scr水平比较 mmol·L-1
大鼠肾脏组织切片HE染色结果:正常组和缺血2 h 组肾小球形态规则,肾小囊整齐紧密排列,肾小管无变形,结构完整;缺血4 h和6 h组可见肾脏肾小球血管袢增生,肾小管轻度水肿扩张,部分区域结构受损。见图1。上述结果说明脑组织缺血时间与AKI密切相关,缺血时间越长肾脏损伤相对越重。
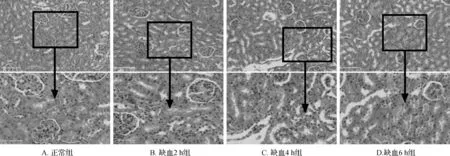
图1 各组大鼠肾脏组织HE染色结果 染色视野均取自肾组织皮质区。A、B、C、D的上图为×200,标尺100 μm; 下图为×400,标尺50 μm
因缺血4 h可见明显肾脏质量指数和肾功能改变,且HE染色可见明显病理改变,故选定缺血4 h为本研究缺血时间。
2.2 不同再灌注时间脑组织病理学变化
在缺血4 h基础上,将再灌注时间点分别设为再灌注3、6、12、24及48 h。脑组织HE染色结果:正常组脑组织结构正常,神经细胞排列整齐,胞质丰富,核仁清晰可见;缺血/再灌注组损伤侧淡染,皮质区胶质纤维溶解、呈筛网状改变,部分缺血边缘区可见液化性坏死,局部可见散在的红细胞和炎症细胞浸润。见图2。上述结果提示脑IR模型构建成功,可进一步行肾脏组织HE染色。
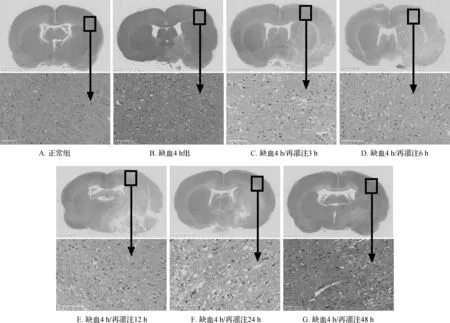
图2 各组大鼠脑组织HE染色结果 A~G的上图为整个脑组织视交叉后2~3 mm切片,标尺2.5 mm; 下图为×200,标尺100 μm
2.3 脑缺血再灌注各时间点肾组织结构变化
肾脏组织HE染色显示,正常组肾脏组织结构完好。缺血4 h/再灌注3 h、缺血4 h/再灌注6 h及缺血4 h/再灌注12 h组均有轻度损伤。但缺血4 h/再灌注24 h和缺血4 h/再灌注48 h组损伤较重,可见肾脏结构分布错乱,部分肾脏皮质片段呈点片状坏死,出现小管上皮细胞萎缩,小管管腔扩张,且伴有局部炎症细胞浸润。见图3。

图3 各组大鼠肾脏组织HE染色结果 A~G的上图为肾组织横切面切片,标尺2.5 mm; 下图为×200,标尺100 μm
结合上述脑组织及肾组织染色结果,选择缺血4 h/再灌注24 h作为本次实验模型的时间点。
2.4 脑缺血再灌注各时间点肾组织纤维化改变
Masson染色结果显示,正常组形态正常,结构完整;在缺血4 h/再灌注24 h时可见肾小球周围及肾小管间质有蓝色胶原沉积,间质纤维组织增生明显;其余各组与正常组之间无明显区别。见图4。上述结果说明缺血4 h/再灌注24 h可引起肾脏胶原沉积,导致肾脏纤维化。

图4 各组大鼠肾脏组织胶原、纤维沉积情况 ×200,标尺100 μm
2.5 IR后各时间点炎症因子水平变化
缺血4 h/再灌注24 h与正常组比较,大鼠血浆炎症因子TNF- α和IL- 6水平明显升高(P<0.05),见表2。
表2 各组大鼠炎症因子TNF- α和IL- 6水平比较 mmol·L-1
2.6 肾脏组织TGF- β1、Smad7及胶原蛋白Ⅰ、Ⅲ mRNA表达的变化
如图5所示,与正常组及缺血4 h组相比,缺血4 h/再灌注24 h组TGF- β1及胶原蛋白Ⅰ、Ⅲ mRNA表达显著增加(P<0.05),而Smad7 mRNA表达显著降低(P<0.05)。
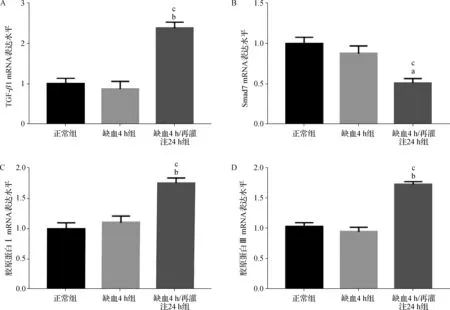
a 与正常组比较,P<0.05; b 与正常组比较,P<0.01; c 与缺血4 h组比较,P<0.01
2.7 肾脏组织TGF- β1、Smad7蛋白表达的变化
蛋白质免疫印迹法检测结果如图6所示。与正常组相比,缺血4 h组TGF- β1表达增加(P=0.007),Smad7

图6 各组大鼠肾脏组织 TGF- β1、Smad7蛋白表达的蛋白质免疫印迹法检测结果
3 讨 论
在临床上可见较多多重器官损伤的病例。机体作为一个整体,一个器官的损伤可以牵动其他器官而产生“综合征”,如肝肾综合征、心肾综合征,此外肺肾综合征和脑肾综合征等也有描述[6- 8]。脑IR被认为不仅会造成脑组织损伤,还会引起远处器官包括肾脏的损伤。有文献报道AKI是急性卒中后的常见并发症,并证明AKI是急性卒中后早期和长期死亡率的独立预测因子[9]。Khatri等[10]观察到,急性缺血性卒中会导致肾功能障碍,并且与住院时间延长和死亡率增高有关。AKI因其较高的死亡率、发病率及昂贵的医疗费用,成为持续性临床问题[11]。AKI可引起肾脏内的炎症反应和细胞凋亡[12],炎症是AKI进展的主要因素;急性炎症反应的特征是炎症细胞的激活和促炎细胞因子的过度分泌。有研究[13]表明,急性脑损伤可能导致AKI,并引发肾脏炎症级联反应。

表3 缺血及再灌注对大鼠肾组织中TGF- β1、Smad7蛋白表达的影响
在本研究中,检测大鼠脑缺血后肾脏功能的指标和HE染色结果表明,大鼠脑缺血4 h时可见血BUN、Scr明显升高,HE染色见肾小管水肿扩张,并且随着脑缺血时间延长肾脏损伤不断加重。明确缺血时间后,通过HE染色筛选不同再灌注时间点大鼠肾脏的病理改变,可见在缺血4 h/再灌注24 h时出现肾脏组织局部炎症浸润,部分皮质片段呈片状坏死。为了探究脑IR后大鼠肾功能出现损伤的机制,进一步行肾组织的Masson染色,可见在缺血4 h/再灌注24 h时即可出现肾脏皮质区胶原沉积,考虑因肾脏纤维化导致,于是检测经典纤维化通路TGF- β1/Smad7相关因子的分子水平改变[14]。炎症细胞的活化聚集、细胞外基质的合成与沉积,二者共同促进了肾小管上皮细胞间充质转化(EMT)改变[15]。Smad7蛋白是TGF- β1受体的胞内激酶底物,是该信号通路的重要介导物质。大量实验证明Smad7作为内源性TGF- β1拮抗剂,可竞争结合TGF- β1受体。此外,Smad7还可诱导TGF- β受体1的泛素- 蛋白酶体降解,从而对TGF- β1/Smad7通路产生负性调节作用[16]。在本研究中,TGF- β1在脑组织IR的动物模型中呈高表达,而其负性调节蛋白Smad7在模型中表达降低;与对照组相比,在脑IR组的肾脏切片中发现炎症细胞浸润增加,且伴有胶原纤维沉积。关于TGF- β1/Smad7 通路激活的原因目前少有报道,检测大鼠脑组织IR后血浆中促炎因子表达,可见呈明显的表达上升趋势,也验证了脑IR后对全身炎症反应系统的激活,从而造成远端脏器的损伤[17]。在今后研究中,可针对TGF- β1这一靶点进行调节,有望改善脑IR引起肾脏纤维化的发生发展。
综上所述,大鼠脑缺血4 h/再灌注24 h时,肾脏组织中TGF- β1/Smad7通路激活,TGF- β1表达明显上升,Smad7表达明显下降,胶原蛋白Ⅰ、Ⅲ表达明显上升,且肾脏组织染色中可见明显胶原沉积现象,出现肾脏纤维化改变。在后续的研究中可通过调节这一通路以减少炎症细胞浸润、减轻氧化应激对肾脏造成的损伤,从而改善肾脏受损。本研究为脑IR损伤诱发AKI等相关疾病的临床研究提供了实验基础。
