气相分子吸收光谱法在地表水氨氮监测中的应用
2022-04-24陈志强费金岩刘京许秀艳王浩
陈志强,费金岩,刘京,许秀艳*,王浩
(1.辽宁省抚顺生态环境监测中心,抚顺 113000;2.中国环境监测总站,北京 100012)
氨氮(NH3-N)是指以游离氨(NH3)或者铵盐(NH4+)形式存在于水体中的氮。两者的组成比取决于水的酸度和温度。当水的酸度偏低,则游离氨比例较高,反之,则铵盐比例较高;水温的影响则相反。水中氨氮的来源主要是生活污水中含氮有机物受微生物作用分解的产物、某些工业废水及农田排水等。此外,死亡的鱼虾以及残余饲料等在细菌的作用下也逐渐分解产生氨。氨氮含量较高时,对鱼类有毒副作用,对人体也有不同程度的危害。因此,氨氮含量的多少是判断水体污染程度的重要标志之一,也是地表水环境质量监测工作中的必测项目。
目前,环境保护部发布的国家环境保护标准HJ 535-2009《水质 氨氮的测定 纳氏试剂分光光度法》中将纳氏试剂分光光度法规定为氨氮的测定方法[1]。该方法灵敏度高,但其测定的水体干扰因素较多,如悬浮物、余氯、钙、镁、硫化物和有机物。当絮凝沉淀不能去除全部干扰时,需采用预蒸馏的前处理方式,操作较为复杂。环境保护部制定的“十三五”国家地表水环境质量监测网设置方案中共设置2 767个断面,“十四五”计划中将进一步调整并增加至3 646个断面,在现有监测任务情况和采测分离的监测模式背景下,仅靠人工操作测试水样压力较大,对于实验室自动仪器分析技术代替纯手工操作方法的需求愈加迫切。
气相分子吸收光谱法是20世纪70年代兴起的一种简便、快速的分析手段,是利用基态的气体分子吸收特定紫外光谱进行定量的一种测定方法,利用该方法进行水中氨氮的测试已有研究报道,并取得了较好的应用效果[2-7]。随着仪器技术的日趋完善,相关国家标准HJ/T 195-2005《水质 氨氮的测定气相分子吸收光谱法》和团体标准T/CHES 15-2017《水质 总氮的测定 气相分子吸收光谱法》应运而生。但气相分子吸收光谱法作为一种相对较新、不断探索进步的方法,其水样测定干扰因素的研究还不全面,水样的保存试验研究也鲜有报道。本工作基于环境保护部制定的“十三五”国家地表水环境质量监测网设置方案的实际监测需要,探究了亚硝酸盐、Ca2+、Mg2+、I-、硫化物对测定结果的影响,考察了气相分子吸收光谱法测定地表水中氨氮时水样保存条件和方法性能指标,并以预蒸馏-纳氏试剂分光光度法的手工测定值为参考,开展了气相分子吸收光谱法的标准样品和实际样品比对分析,以展望该方法在地表水环境质量监测中的应用前景。
1 试验部分
1.1 仪器与试剂
AJ-3700型气相分子吸收光谱仪;TU-1901 型紫外-可见分光光度计;STEHDB-106-3型智能一体化蒸馏仪。
纳氏试剂:称取氢氧化钠8.0 g溶于50 mL 水中,冷却至室温。称取碘化钾3.5 g和碘化汞5.0 g溶于水中,将此溶液在搅拌下缓慢加入到上述氢氧化钠溶液中,用水稀释至100 mL。贮存于聚乙烯瓶内,用橡皮塞或聚乙烯盖子盖紧,在暗处可保存1 a。
酒石酸钾钠溶液:500 g·L-1,称取50.0 g酒石酸钾钠溶于100 mL水中,加热煮沸3~5 min,冷却至室温后用水稀释至100 mL,摇匀,备用。
盐酸-乙醇溶液:分别移取300 mL 盐酸和250 mL无水乙醇,用水稀释并定容至1 L,充分摇匀,静置2 h以上备用,作为气相分子吸收光谱法中的载流液。
溴酸盐混合储备溶液:称取2.81 g 溴酸钾及30 g溴化钾,溶解于500 mL 水中,摇匀,储存于玻璃瓶中。
次溴酸盐溶液:移取6.0 mL 溴酸盐混合储备溶液置于棕色磨口试剂瓶中,加入水200 mL 及6 mol·L-1盐酸溶液12.0 mL,立即密塞,充分摇匀,于暗处放置10~20 min,加入100 mL 40%(质量分数)氢氧化钠溶液,充分摇匀,待小气泡逸尽再使用。该溶液临用时配制,作为气相分子吸收光谱法中的氧化剂。
溴酸钾、溴化钾、氯化铵、轻质氧化镁、溴百里酚蓝、无水乙醇、氢氧化钠、硫酸、盐酸均为分析纯;试验用水为无氨水。
1.2 试验方法
1.2.1 样品采集保存及前处理
1)采集保存 采集地表水水样加入具塞玻璃瓶中,立即加入硫酸使水样酸化至p H<2,固定、运输及保存过程中始终加盖密闭。采样后尽量在24 h内进行测定。
2)预蒸馏法 将50 mL 无氨水吸收液移至250 mL容量瓶内,将冷凝管出口插入容量瓶中,确保冷凝管出口在无氨水吸收液液面之下。用量筒分取250 mL 水样(如氨氮含量高,可适当少取,加水至250 mL)移至烧瓶中,加几滴溴百里酚蓝指示剂,必要时,用氢氧化钠溶液或盐酸溶液调整pH 至6.0(指示剂呈黄色)~7.4(指示剂呈蓝色),加入0.25 g轻质氧化镁,连接好管路。加热蒸馏,使馏出液速率约10 mL·min-1,待馏出液达200 mL 时,停止蒸馏,用水定容至250 mL。
1.2.2 气相分子吸收光谱法
气相分子吸收光谱法参照HJ/T 195-2005。将载流液、氧化剂连接到对应的进液管上,配制2.0 mg·L-1的氨氮标准溶液,放置在进样盘上,启动AJ-3700软件工作站,选择测定项目“氨氮”,测定方式“峰高”,氘灯电流为50 m A,标准测定方式“单标准定标法”,以空气为载气,其他参数为仪器默认设置,在波长214.7 nm 处测量吸光度,绘制氨氮质量浓度及其对应吸光度的标准曲线。将适量的水样倒入进样管,放置在样品盘上,按照上述步骤进行氨氮测定。
1.2.3 纳氏试剂分光光度法
纳氏试剂分光光度法参照HJ 535-2009。移取10.0 mg·L-1氨氮标准溶液0,0.5,1.0,2.0,4.0,6.0,8.0,10.0 mL 置于50 mL 的比色管中,加水 至刻度。加入1.0 mL 酒石酸钾钠溶液,摇匀,再加入纳氏试剂1.0 mL,摇匀。放置10 min后,用20 mm比色皿,以水作参比,于波长420 nm 处测量吸光度,绘制氨氮质量浓度及其对应吸光度的标准曲线。取适量的水样置于50 mL 的比色管中,加水至刻度,按上述相同步骤测量吸光度,同时作空白测定,然后手工计算氨氮含量。
2 结果与讨论
2.1 样品保存试验
在水样氨氮的实际监测工作中,样品稳定性是值得关注的重要环节。在样品的保存过程中,水中的氨氮有可能发生转化,使测定结果偏离实际。只有采用合理的保存方法,并在有效的保存时间内进行及时分析,才能获得可靠的分析数据。以6个有检出的实际水样(样品31~36)为研究对象,在4 ℃冷藏条件下,试验考察了加酸前后保存时间对样品测定结果的影响,以新鲜水样测定值为基准计算相对偏差,结果见图1。

图1 加酸前后样品保存时间对氨氮测定的影响Fig.1 Effect of storage time of the sample on determination of ammonia nitrogen before and after adding acid
由图1可知:样品32在加酸及不加酸时7 d稳定性均较高,测定结果相对偏差均在5%以内;样品36加酸时7 d内的相对偏差控制在5%以内,而不加酸时在7 d的相对偏差超过10%;样品34和31加酸时14 d的相对偏差均小于10%,而不加酸时7 d的测定结果相对偏差则超过50%;样品33加酸保存7 d的相对偏差小于20%,不加酸保存7 d的相对偏差达到100%;样品35加酸保存7 d的相对偏差小于20%,但不加酸保存1 d的相对偏差则超过90%。综上所述,加酸后样品的稳定性明显高于未加酸时的稳定性,加酸保存,7 d内样品测定结果的相对偏差均小于20%。特别对于相对敏感,不易保存的样品,如样品33和样品35,不加酸保存时,水样甚至在1 d内其测定结果就明显降低。综合考虑我国地表水基体的多样性和复杂性,在不加酸固定剂的情况下,应尽量在1 d内完成水样分析;即使在加酸固定的情况下,也应在7 d内需完成水样分析,以保证监测数据的准确性。
2.2 亚硝酸盐对氨氮测定的影响
根据气相分子吸收光谱法测定氨氮的原理,氨及铵盐先被氧化成亚硝酸盐后再转化成二氧化氮被测定,因此亚硝酸盐对氨氮的测定存在正干扰。试验对含有不同质量浓度亚硝酸盐的空白水样和同时含有1.0 mg·L-1氨氮和不同质量浓度亚硝酸盐的溶液进行分析,氨氮测定结果随亚硝酸盐质量浓度变化的曲线见图2。
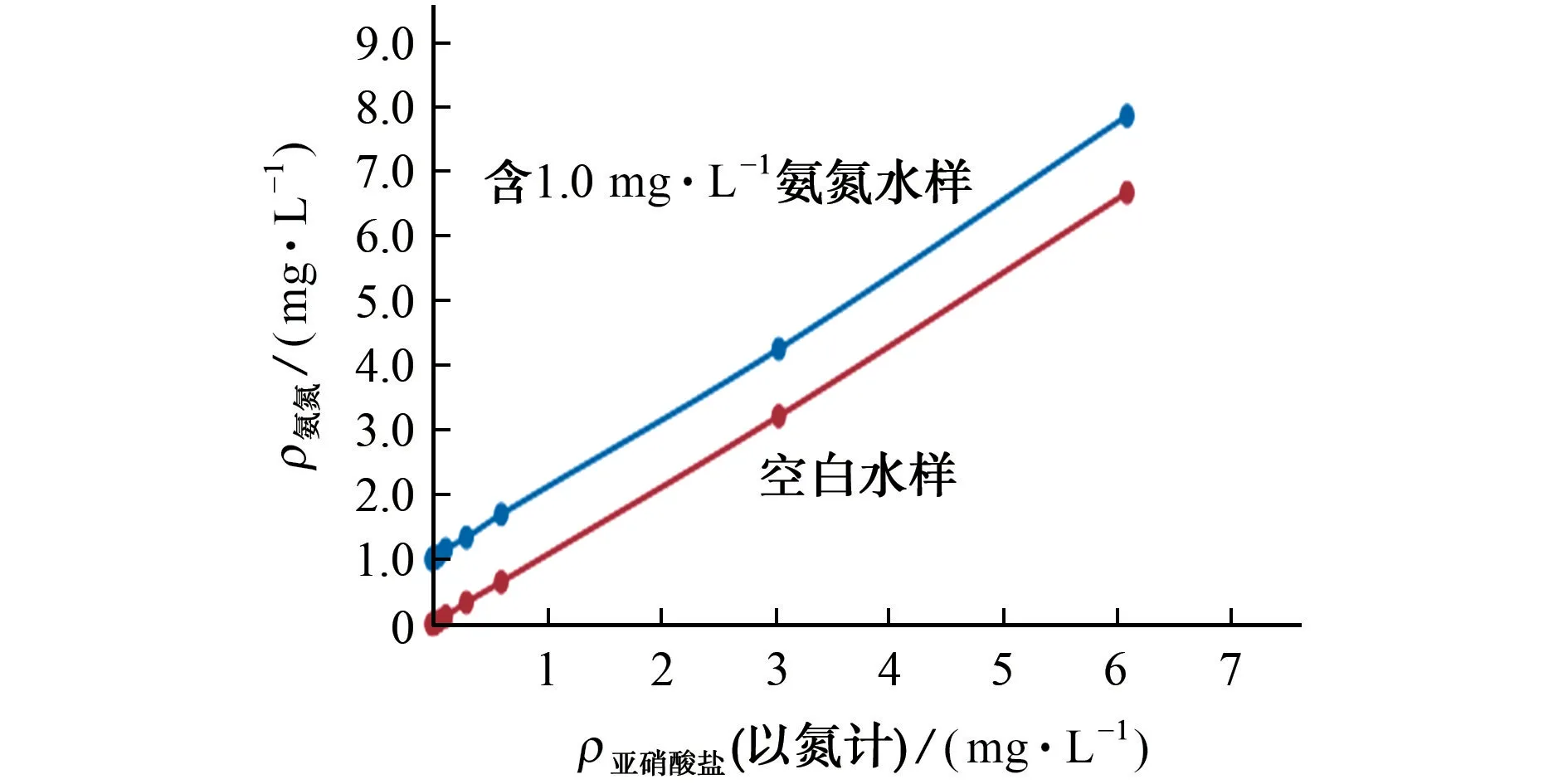
图2 亚硝酸盐对氨氮测定的影响Fig.2 Effect of nitrite on determination of ammonia nitrogen
由图2 可知,无论是空白水样,还是含有1.0 mg·L-1氨氮的水样,其氨氮测定结果均随着样品中亚硝酸盐质量浓度(以氮计)的增加而增加,并且测定结果的相对误差和亚硝酸盐的质量浓度基本相当。
当水样中含有亚硝酸盐时,水样分析时可选用仪器操作系统中的氨氮除亚氮功能模式,当启用该模式时,含1.0 mg·L-1氨氮水样中氨氮测定结果随亚硝酸盐质量浓度变化的曲线见图3。
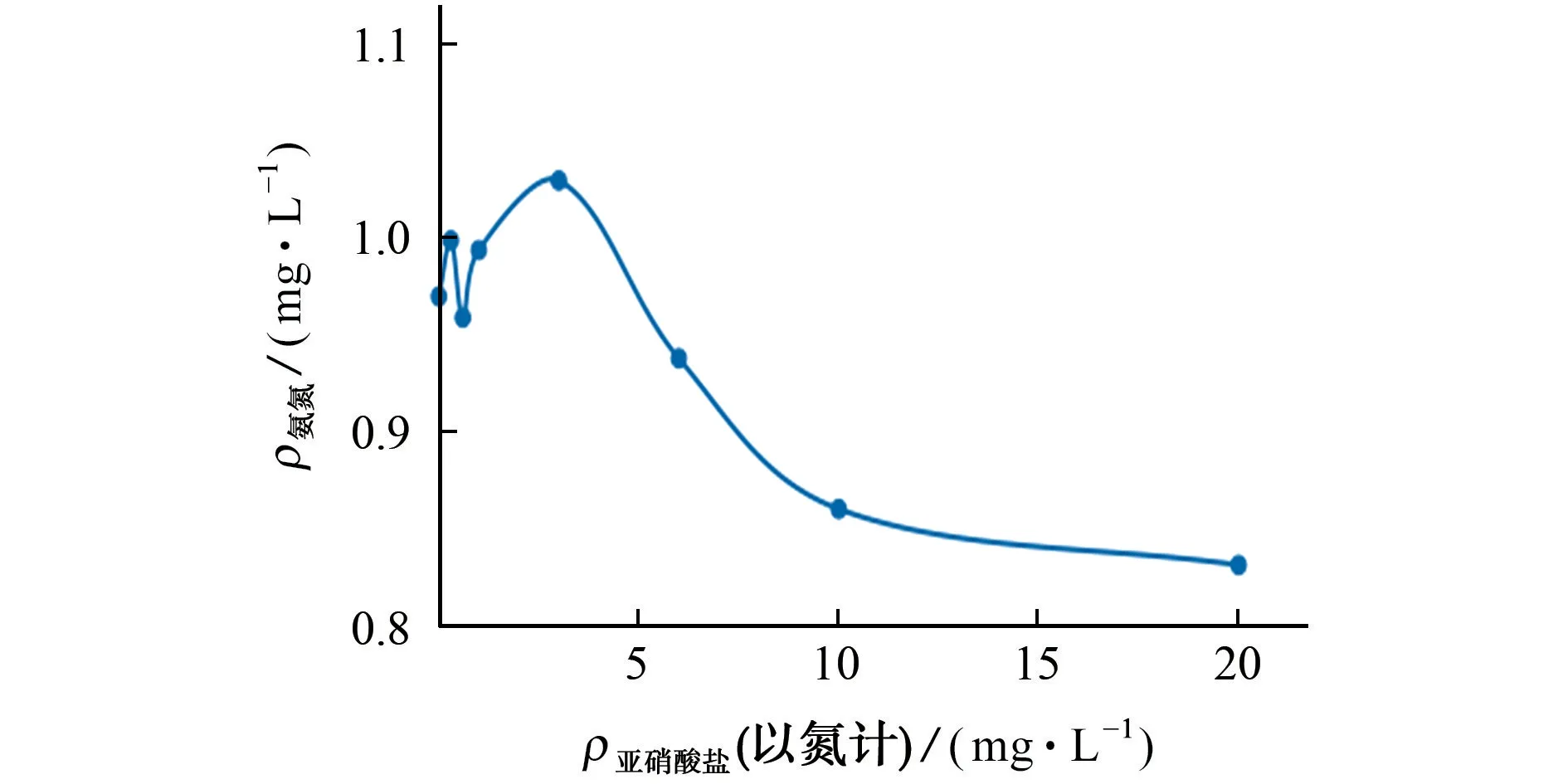
图3 氨氮除亚氮功能模式下亚硝酸盐对氨氮测定的影响Fig.3 Effect of nitrite on determination of ammonia nitrogen under the fuction mode of ammonia nitrogen removal of nitrite
结果表明,当亚硝酸盐质量浓度(以氮计)小于3 mg·L-1时,含1.0 mg·L-1氨氮水样的测定相对误差绝对值在5.0%以内;随着亚硝酸盐质量浓度的增大,水样测定结果的相对误差明显增加,选用氨氮除亚氮功能模式已不能满足干扰的消除要求,在水样分析前,只有采用加热煮沸或预蒸馏前处理方式才能有效消除亚硝酸盐的干扰。
2.3 Mg2+、Ca2+对氨氮测定的影响
采用纳氏试剂分光光度法测定水中氨氮时,当水中存在Mg2+、Ca2+时,在碱性溶液中易水解产生沉淀,影响测定结果。表1 给出了纳氏试剂分光光度法和气相分子吸收光谱法测定含1.0 mg·L-1氨氮和不同质量浓度Mg2+、Ca2+的水样的测定结果。
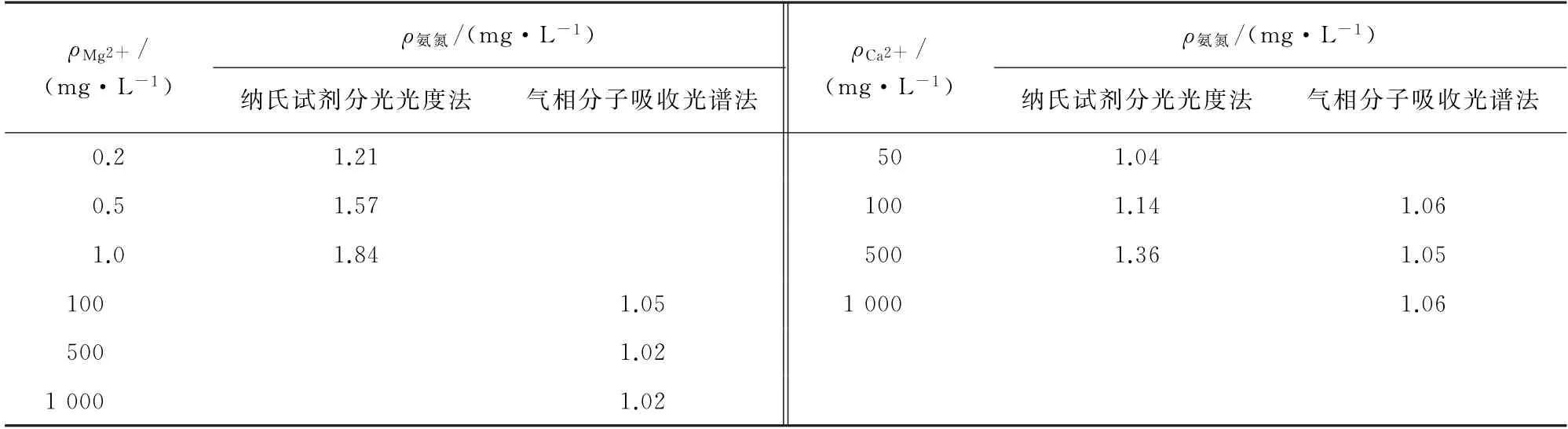
表1 Mg2+、Ca2+对氨氮测定的影响Tab.1 Effect of Mg2+and Ca2+on determination of ammonia nitrogen
结果表明:当水样中Ca2+超过100 mg·L-1或Mg2+超过0.2 mg·L-1时,采用纳氏试剂分光光度法获得的测定结果发生明显偏差,需加入酒石酸钾钠溶液进行掩蔽;而基于气液分离原理的气相分子吸收光谱法测定结果完全不受水中Mg2+、Ca2+的影响。这进一步说明在测定高硬度水体中的氨氮时,气相分子吸收光谱法比纳氏试剂分光光度法更具有优势。
2.4 I-对氨氮测定的影响
团体标准T/CHES 15-2017中指出气相分子吸收光谱法适用于海水的测定。因此,试验进一步考察了I-对氨氮测定的影响。图4 给出了含1.0 mg·L-1氨氮水样的测定结果随I-质量浓度变化的曲线。
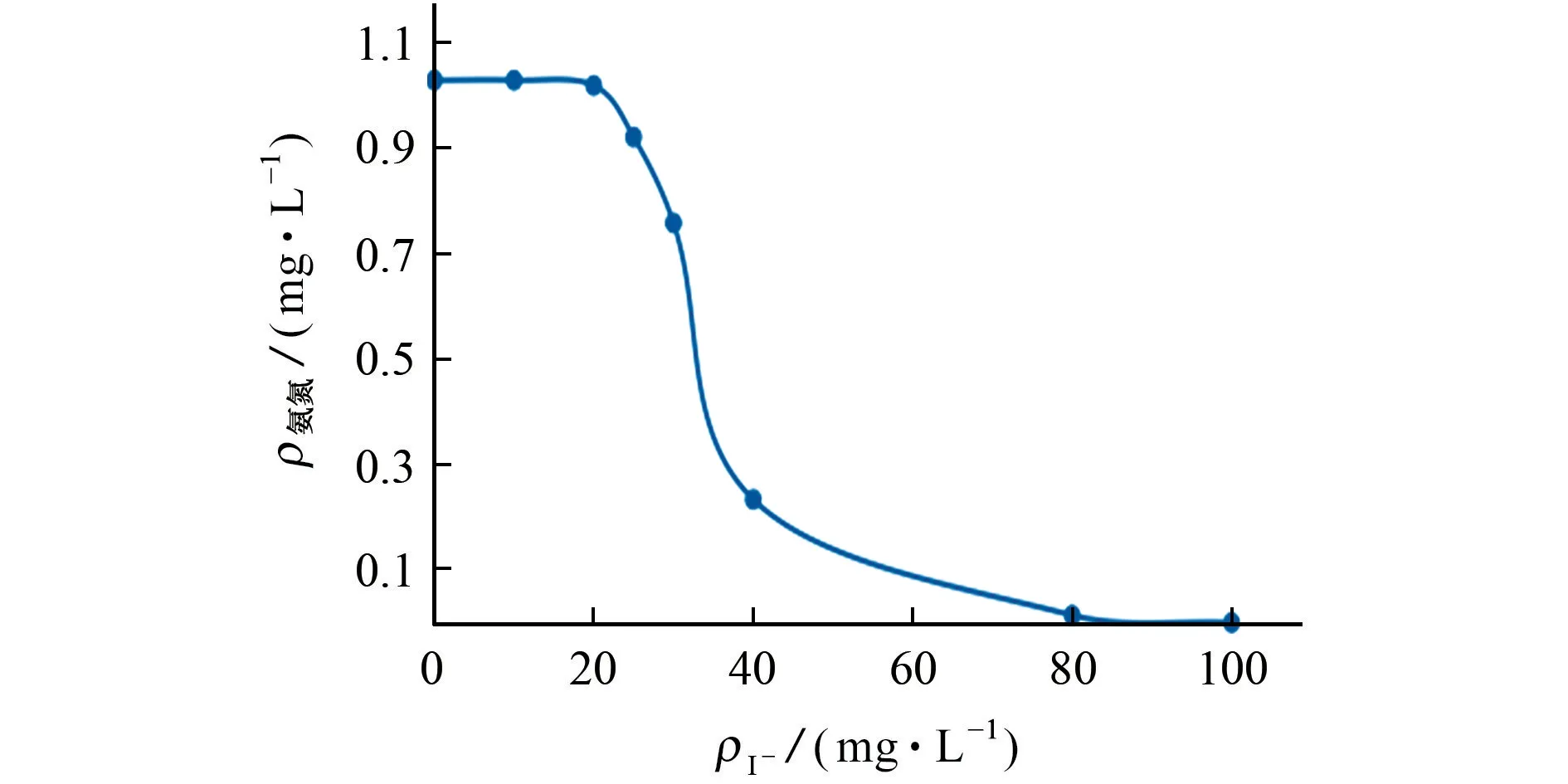
图4 I-对氨氮测定的影响Fig.4 Effect of I-on determination of ammonia nitrogen
结果表明,水样的测定结果随着I-质量浓度的增加而降低,这是因为在酸性条件下,氧化剂次溴酸的标准电极电位为1.33 V,大于I-的(0.99 V)而小于Cl-的(1.50 V),次溴酸可以将I-氧化,随着I-质量浓度的加大,氧化剂的质量浓度相对逐渐减少,从而影响到氧化剂与氨氮的反应,致使氨氮的测定结果偏低。但是,该影响相对较小,当I-的质量浓度达到25 mg·L-1时,测定结果的相对误差也仅达到7.8%,而通常海水中碘的平均质量分数为0.05 mg·kg-1[8],因此在实际环境监测中,只有遇到高碘废水时才需考虑I-的干扰。
2.5 硫化物对氨氮测定的干扰及消除
硫化物的标准电极电位为0.141 V,次溴酸可氧化硫化物,因此硫化物对氨氮的测定存在干扰。试验对含有不同浓度硫化物的空白水样和同时含1.0 mg·L-1氨氮的水样进行分析,氨氮测定结果随硫化物质量浓度变化的曲线见图5。

图5 硫化物对氨氮测定的影响Fig.5 Effect of sulfide on determination of ammonia nitrogen
由图5可知,当硫化物质量浓度低于10 mg·L-1时,含1.0 mg·L-1氨氮水样测定结果的相对误差绝对值小于5.0%,此时硫化物对氨氮的测定基本无干扰,但随着硫化物质量浓度的增加,氨氮的测定结果下降到一定值后又逐渐升高。原因可能为:当硫化物质量浓度在10 mg·L-1以内时,硫化物虽与试验过程中的部分氧化剂发生反应,但剩余的氧化剂仍然满足与氨氮的反应要求,因此测定结果不受影响;当硫化物质量浓度为10~20 mg·L-1时,硫化物消耗了过多氧化剂,导致氨氮氧化反应不完全,使得测定结果偏低;当硫化物质量浓度大于20 mg·L-1时,氧化剂仅能与部分硫化物发生反应,剩余的硫化物与试验中的盐酸反应生成硫化氢,硫化氢对氨氮的测定造成正干扰,使得测定结果偏高。在实际地表水监测工作中,这种高质量浓度硫化物的水样几乎不存在,因此硫化物的干扰影响基本可忽略。
2.6 检出限
根据HJ 168-2010《环境监测 分析方法标准制修订技术导则》中规定的计算检出限的方法[9],采用气相分子吸收光谱法测定空白加标样品(加标量0.05 mg·L-1),连续测定7次,按照t×s计算方法的检出限。当置信水平为99%时,t值为3.143,s为测定7次的标准偏差,则检出限(3.143s)结果为0.02 mg·L-1。
2.7 精密度和回收试验
采用气相分子吸收光谱法测定空白加标样品(加标量0.20,0.80,1.60 mg·L-1),按试验方法处理,每个水样平行测定6次,计算回收率和测定值的相对标准偏差(RSD),结果见表2。

表2 精密度和回收试验结果(n=6)Tab.2 Results of tests for precision and recovery(n=6)
由表2可知,氨氮回收率为94.7%~101%,测定值的RSD 为0.70%~4.7%。
2.8 标准样品和实际样品的数据比对分析
目前,环境保护部制定的“十三五”国家地表水环境质量监测网设置方案对水样氨氮的测定方法统一规定为纳氏试剂分光光度法,前处理方式为絮凝沉淀或预蒸馏。考虑到实际水样的复杂性,为避免水样基体可能带来的干扰,试验以预蒸馏-纳氏试剂分光光度法的手工测定值为参考,开展了气相分子吸收光谱法的标准样品和实际样品比对分析。
2.8.1 标准样品的比对分析
选取3 个典型质量浓度的氨氮标准样品(2005102,206912,200559)进行了方法的比对分析,结果见表3。

表3 标准样品的不同方法比对结果Tab.3 Results of comparison by different methods for standard samples
由表3可知,采用气相分子吸收光谱法分析标准样品,其测定结果均在标准样品认定值的允许偏差范围内。采用纳氏试剂分光光度法分析标准样品,整体结果偏低,只有高质量浓度标准样品的测定结果在标准样品认定值的允许偏差范围内。分析原因可能是标准样品无基体干扰,定值时通常不经前处理直接测定,而当低质量浓度标准样品经预蒸馏前处理后测定时,预蒸馏过程硼酸吸收不完全或者后续显色干扰,均可能导致测定结果偏低。另外,由气相分子吸收光谱法两种前处理方式数据对比可知,预蒸馏前处理对测定结果基本无影响,说明纳氏试剂分光光度法结果偏低主要归因于显色环节硼酸对纳氏试剂的干扰,文献[10-11]亦有报道。而且,由表3中相对偏差数据可以看出,气相分子吸收光谱法与纳氏试剂分光光度法测定标准样品的相对偏差的绝对值在10%以内,同时对气相分子吸收光谱法与纳氏试剂分光光度法得到的测定结果进行显著性检验,得到P>0.05,说明两种方法测定结果无显著性差异。
2.8.2 样品比对分析
选取了不同浓度水平的国控、省控断面的地表水样品,开展了比对分析,结果见表4。
由表4可知,当水体中氨氮质量浓度低于V 类标准限值水平(2.0 mg·L-1)[12]时,纳氏试剂分光光度法测定的结果通常低于气相分子吸收光谱法测定的结果,这主要是由硼酸在显色反应时的干扰造成,前面已有赘述。对于高质量浓度水样,综合基体的复杂性和两种方法原理的差异性,测定结果的高低并无明显规律。而对于气相分子吸收光谱法来说,直接进样分析与预蒸馏后分析的结果基本一致,仅有个别水样直接进样的测定结果明显高于预蒸馏后的结果,这主要是由复杂基体的干扰造成。因此,在选择气相分子吸收光谱法测定水样中的氨氮时,对于复杂水体,分析前必须经过预蒸馏处理。

表4 样品的不同方法比对结果Tab.4 Results of comparison by different methods for samples
本工作采用预蒸馏-气相分子吸收光谱法测定水样中氨氮的含量,具有较好的精密度和准确度。在标准样品和实际水样的分析研究中,与纳氏试剂分光光度法具有较好的可比性,而且该方法操作简单、自动化程度高,适于样品的批量分析,在我国环境监测领域具有广阔的应用前景。
