π-π堆积的氧化还原型阿霉素脂质体的制备与抗肿瘤活性评价
2022-04-24孙东起郑海涛刘钧华姚子涵祝艳平张雷明凌龙兵
单 琪,孙东起,郑海涛,刘钧华,姚子涵,祝艳平,杜 源,张雷明,凌龙兵
(烟台大学药学院,新型制剂与生物技术药物研究山东省高校协同创新中心,长效和靶向制剂国家重点实验室,山东 烟台 264005)
阿霉素(Doxorubicin,DOX)是目前临床上最常用的一线抗肿瘤药物,具有显著抗肿瘤活性。DOX主要在肝脏代谢,其骨髓抑制和心脏毒性较大,长期使用可引起严重的心脏毒性和肝脏损害,导致临床应用受限[1-3]。脂质体作为近十几年来被广泛关注的纳米载体,在肿瘤的化学治疗、基因治疗等方面的靶向递送中发挥着重要作用[4-5]。以传统脂质材料(包括甘油磷脂,鞘磷脂等)为载体,药物选择性吸附或包裹于脂质双分子层或脂质核中,形成纳米脂质体给药系统,具有高生物相容性和低毒性。但传统脂质体因其双相释药特性,前期突释,后期缓释,无法实现在靶部位的敏感释放且载药量低(<10%)、稳定性差,这可能是脂质纳米载药体系并没有良好的体内抗肿瘤活性的原因[6-8]。到目前为止,还没有非常有效的方法从新型“磷脂材料设计”层面根本上解决这一难题。本课题设计合成新型“功能化二倍体磷脂材料”,在传统磷脂材料碳骨架基础上,内嵌吡啶基与双硫键于磷脂硬脂酸链合成功能化磷脂Pyr-SS-PC,并用其构建π-π堆积的DOX脂质体,结构层面赋予传统载药脂质体的“高载药量和促释放”功能。考察DOX包封率及体外响应释药行为,构建乳腺癌细胞MCF-7荷瘤裸鼠模型,研究DOX/Pyr-SS-PC Lips对实体瘤的肿瘤抑制率,为进一步研究脂质体剂型积累实验基础。
1 仪器与材料
1.1 仪器
Ultimate 3000高效液相色谱仪(美国Thermo Fisher公司);Q Exactive液相色谱-高分辨质谱联用仪(美国Thermo Fisher公司);UltraShield核磁共振仪(瑞士Bruker公司);RE-2000B旋转蒸发仪(上海亚荣生化仪器厂);Zetasizer Nano ZS90纳米粒度电位仪(英国马尔文公司);SpectraMax M3全波长扫描酶标仪(美国MD公司);JEOL JEM-1230透射电子显微镜(日本电子株式会社)。
1.2 材料
阿霉素(北京中硕医药科技开发有限公司,批号J0614A,纯度> 98%);大豆卵磷脂、胆固醇(上海麦克林生化科技有限公司,纯度> 99%);DSPE-PEG2000(西安瑞禧生物科技有限公司);巯基十一烷酸、三苯基氯甲烷、二硫二吡啶、N,N'-羰基二咪唑(CDI)和1,8-二氮杂二环[5.4.0]十一碳-7-烯(DBU)均购于上海阿拉丁生化科技股份有限公司;RPMI-1640培养基、胎牛血清(美国Gibco公司);葡聚糖凝胶G50(上海笛柏生物科技有限公司);透析袋(MWCO 3500)(Sigma-Aldrich,美国);其他试剂均为市售分析纯。MCF-7乳腺癌细胞株、HeLa宫颈癌细胞株和HepG-2肝癌细胞株(中国医学科学肿瘤细胞库,自行传代);Balb/c裸鼠,SPF级,雌性,体重18~20 g。
2 方 法
2.1 Pyr-SS-PC的合成
Pyr-SS-PC磷脂的合成主要包括三步:(1)三苯基醚保护巯基十一烷酸Tri-S-COOH;(2)在CDI/DBU强催化条件下,Tri-S-COOH与GPC反应生成双三苯基醚保护十一巯基烷酸甘油磷脂酰胆碱(Tri-S-PC);(3)脱保护的Tri-S-PC经巯基-二巯基交换反应得到吡啶基双硫甘油磷脂酰胆碱(Pyr-SS-PC)。具体的反应路线如图1所示。
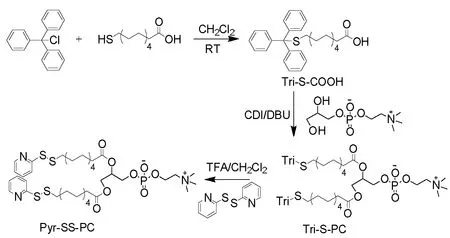
图1 吡啶基双硫磷酸胆碱Pyr-SS-PC合成路线
2.1.1 Tri-S-COOH的合成 将1 g(4.58 mmol)巯基十一烷酸溶解于20 mL无水二氯甲烷中,搅拌中逐滴加入2.55 g(9.16 mmol)三苯基氯甲烷的二氯甲烷溶液,室温反应6 h。反应结束后,浓缩反应液,用200 mL石油醚重结晶2次,得到白色固体2.01 g,收率95.2%。1H NMR (500 MHz, CDCl3):δ7.36~7.23 (m, 9H), 2.59 (s, 1H), 2.27 (s, 1H), 1.63 (s, 1H), 1.47 (s, 1H), 1.40 (d,J=7.7 Hz, 2H), 1.31 (dd,J=18.0, 5.5 Hz, 5H);13C NMR (125 MHz, CDCl3):δ177.13 (s, 1H), 145.34 (s, 7H), 129.38 (s, 14H), 129.13 (s, 15H), 127.79 (s, 3H), 64.64 (s, 2H), 34.64 (s, 2H), 30.23 (s, 5H), 28.95 (t,J=1.6 Hz, 15H), 24.81 (s, 2H)。HRMS, ESI+,m/z: Calcd for C30H36O2S [M-H]-459.24, found 459.24。
2.1.2 Tri-S-PC的合成 将0.46 g(0.99 mmol)三苯基醚保护巯基十一烷酸和0.24 g/1.49 mmol CDI溶解于15 mL无水二甲基亚砜中,35 ℃活化2 h。向上述反应体系中,继续加入0.10 g/0.39 mmol 甘油磷酸胆碱和0.23 g(1.49 mmol)DBU,45 ℃条件下反应过夜。反应结束后,用含有10%冰醋酸的乙醚溶液沉降反应液,浓缩后利用三氯甲烷/甲醇/水(体积比65∶25∶4)柱层析,得到0.64 g双三苯基醚保护巯基烷酸甘油磷脂酰胆碱,收率56.4%。1H NMR (500 MHz, CDCl3):δ7.40~7.34 (m, 14H), 7.34~7.25 (m, 16H), 5.43 (s, 1H), 4.51 (s, 1H), 4.29 (d,J=8.0 Hz, 3H), 3.99 (d,J=30.1 Hz, 2H), 3.80 (s, 2H), 3.24 (s, 9H), 2.52 (s, 2H), 2.47~2.38 (m, 6H), 1.74 (s, 1H), 1.73~1.64 (m, 6H), 1.49~1.41 (m, 6H), 1.41~1.13 (m, 35H).13C NMR (125 MHz, CDCl3):δ174.55 (s, 1H), 174.36 (s, 2H), 145.34 (s, 14H), 129.38 (s, 28H), 129.13 (s, 29H), 127.79 (s, 14H), 69.29 (s, 2H), 68.20 (s, 2H), 66.81 (s, 2H), 64.64 (s, 5H), 64.25 (s, 2H), 60.53 (s, 1H), 54.72 (s, 5H), 34.09 (d,J=15.0 Hz, 4H), 30.23 (s, 10H), 29.07~28.74 (m, 47H), 25.33 (s, 5H)。 HRMS, ESI+,m/z: Calcd for C68H88NO8PS2[M+H]+1 142.57, found 1 142.57。
2.1.3 Pyr-SS-PC的合成 将0.2 g(0.17 mmol)双三苯基醚保护巯基烷酸甘油磷脂酰胆碱溶解于10 mL三氟乙酸/二氯甲烷(体积比1∶1)溶液中,室温反应4 h。无需进一步后处理,在50 ℃下,利用旋转蒸发法除去三氟乙酸脱保护试剂。向上述体系中,加入0.15 g(0.68 mmol)二硫二吡啶的二氯甲烷溶液(10 mL),继续室温反应24 h。反应结束后,浓缩后利用三氯甲烷/甲醇/水(体积比65∶25∶4)柱层析,得到0.13 g双巯十一吡啶基甘油磷脂酰胆碱,收率86.3%。1H NMR (500 MHz, CDCl3):δ8.26 (dd,J=7.5, 1.4 Hz, 3H), 7.57~7.52 (m, 3H), 7.35 (dd,J=7.5, 1.4 Hz, 3H), 7.20~7.15 (m, 3H), 5.45 (s, 1H), 4.63~4.42 (m, 2H), 4.42~4.38 (m, 1H), 4.33 (s, 4H), 4.01~3.97 (m, 1H), 3.80 (s, 3H), 3.24 (s, 12H), 2.56~2.52 (m, 5H), 2.50 (s, 3H), 2.42 (s, 3H), 1.69 (d,J=5.9 Hz, 5H), 1.63~1.60 (m, 4H), 1.54 (dd,J=47.8, 3.9 Hz, 2H), 1.60~1.16 (m, 34H).13C NMR (125 MHz, CDCl3):δ174.55 (s, 2H), 174.36 (s, 1H), 160.01 (s, 3H), 145.72 (s, 3H), 139.66 (s, 3H), 121.37 (s, 3H), 119.67 (s, 1H), 69.29 (s, 1H), 68.20 (s, 1H), 66.81 (s, 1H), 64.25 (s, 1H), 60.53 (s, 1H), 54.72 (s, 5H), 38.03 (s, 3H), 34.09 (d,J=15.0 Hz, 3H)。 28.93 (dd,J=6.7, 5.0 Hz, 19H), 25.33 (s, 1H). HRMS, ESI+,m/z: Calcd for C40H66N3O8PS2[M+H]+876.35, found 876.35。
2.2 阿霉素脂质体的制备与表征
利用超声波分散法制备阿霉素脂质体(DOX/Pyr-SS-PC Lips)。分别称取处方量的56 mmol Pyr-SS-PC、35 mmol大豆卵磷脂、38 mmol胆固醇、6 mmol DSPE-PEG2000和6 mmol DOX溶解于15 mL的氯仿-甲醇溶液中(体积比4∶1)。充分振荡,使其混合均匀。在35 ℃减压旋转蒸发30 min除去溶剂,加入15 mL磷酸盐缓冲溶液(PBS,pH=7.4),50 ℃水化孵育1 h。将所得的混悬液置于200 W探头下超声20 min后,依次经过直径800、450和220 nm聚碳酯膜制得DOX/Pyr-SS-PC Lips。
粒径与Zeta电位测定:取上述制备的DOX/Pyr-SS-PC Lips用纳米粒度电位仪(DLS)测定其粒径、粒径分布和Zeta电位。测试条件:温度为25 ℃,平衡时间9 min,散射角173°。每个样品重复测试三次。
形态观察:采用透射电子显微镜(TEM)观察脂质体形貌特征。测试方法:取5 μL DOX/Pyr-SS-PC Lips溶液滴加到300目碳膜支撑的铜网上,晾干后滴加磷钨酸染色液(质量浓度2%)进行负染,滤纸拭去多余染液。室温干燥后用TEM观察拍照。
2.3 阿霉素脂质体的包封率的测定
采用超滤离心-紫外分光光度计法测定脂质体中DOX包封率,检测波长为480 nm。精密移取两份各1 mL的DOX/Pyr-SS-PC Lips,一份置于Millipore Ultra-4超滤管中,4500 r/min离心10 min。收集下清液,用色谱级甲醇稀释10倍,紫外分光光度计测定吸光度值A1;另一份直接用色谱级甲醇稀释10倍,测定其吸光度值A2。按下述公式计算DOX包封率(EE):EE=(A1/A2)×100%[9]。
2.4 π-π堆积作用的表征
利用紫外-可见吸收光谱检测载药纳米脂质体Pyr-SS-PC与DOX之间存在的π-π堆积作用[10]。精确移取0.5 mL的游离DOX甲醇溶液和DOX/Pyr-SS-PC Lips,对比分析实验组的紫外-可见吸收光谱数据,判断载体Pyr-SS-PC与药物DOX的π-π堆积作用。
2.5 体外释药及血浆稳定性
利用动态透析法考察DOX/Pyr-SS-PC Lips体外响应释药行为。向透析袋(MWCO 3500 U)中加入脂质体2 mL,将透析袋置于35 mL的PBS(pH=7.4)或含有10 mmol/L GSH的PBS溶出介质中透析。在37 ℃、100 r/min的恒温振荡器中震荡,于1、2、4、6、12、24、48、72 h 取1 mL透析液测定480 nm下DOX的吸光度,计算DOX脂质体的累积释放量。
取Balb/c昆明小鼠血浆适量,离心得上清液。向1 mL的血清中分别加入100 μL的DOX/Pyr-SS-PC Lips(0.1 mg DOX/mL)和同浓度、同体积的游离DOX溶液,放置于37 ℃中。在预定的时间点,取20 μL试验用色谱级甲醇稀释10倍,利用紫外分光光度计测定DOX于480 nm下吸光度值。
2.6 体外细胞毒性实验
DOX/Pyr-SS-PC Lips的体外细胞毒性采用噻唑蓝(MTT)法进行评价,并以游离盐酸阿霉素水溶液作为阳性对照组[11]。取对数生长期的人乳腺癌细胞MCF-7、宫颈癌细胞HeLa和肝癌细胞HepG-2按1×104个/孔接种于96孔板中,在5% CO2、37 ℃培养箱内孵育过夜。设置不含DOX的RPMI-1640培养基为空白组,向培养板中分别加入不同浓度的游离DOX和DOX/Pyr-SS-PC Lips,每组设置6复孔。于加药后培养24 h,每孔加入20 μL、5 mg/mL MTT溶液继续孵育4 h。弃去上清液,每孔加入150 μL DMSO,利用酶标仪检测490 nm处每孔吸光度(A),吸光度以平均值作为最终结果。计算细胞增殖抑制率:细胞抑制率=[1-(A实验组-A空白组)/(A对照组-A空白组)]×100%。
采用回归方程式计算游离DOX和DOX/Pyr-SS-PC Lips对3株细胞的半数抑制浓度(IC50)。
2.7 体内抗肿瘤活性
(1)荷MCF-7肿瘤裸鼠模型:Balb/c裸鼠饲养在标准清洁级SPF环境中,温度18~25 ℃、湿度40%~60%。收集浓度为1×107个/mL的人乳腺癌MCF-7细胞悬液,并接种于Balb/c裸鼠右侧腋窝皮下,200 μL/只。
(2)给药方式:待肿瘤体积生长至100~150 mm3,随机分为3组,每组4只,按照如下给药方案,尾静脉注射给药。I. 0.9%生理盐水注射液组(0.1 mL/10 g,间隔1 d给药,共给药8次);II.游离DOX注射液组(5 mg DOX/kg,间隔1 d给药,共给药8次);III. DOX/Pyr-SS-PC Lips(5 mg DOX/kg,间隔1 d给药,共给药8次)。
(3)抑瘤率:给药结束后,将各实验组裸鼠经乙醚麻醉后脱颈处死,手术剥取瘤块称重,并计算抑瘤率。抑瘤率=[(给药组平均瘤重-模型组平均瘤重)/模型组平均瘤重]×100%。
2.8 统计学方法
3 结果与讨论
3.1 Pyr-SS-PC的合成及表征
根据图1实验反应路线合成吡啶基双硫Pyr-SS-PC磷脂,并利用HRMS、1H NMR和13C NMR 对各合成中间体与产物进行详细的结构表征。首先,巯基十一烷酸在碱性TEA催化环境下,与巯基保护剂三苯基氯甲烷(TriCl)反应,得到Tri-S-COOH;之后,Tri-S-COOH与甘油磷酸胆碱(GPC)在CDI/DBU强催化条件下,生成Tri-S-PC偶联物。最后,通过巯基—二巯基交换反应,二硫二吡啶与巯基脱保护的Tri-S-PC反应以获得产物吡啶基双硫甘油磷脂酰胆碱Pyr-SS-PC。
利用HRMS、1H NMR和13C NMR对Pyr-SS-PC的精准化学结构进行表征(图2)。

图2 Pyr-SS-PC磷脂结构表征
如图2(a)所示,Pyr-SS-PC产物分子离子峰为876.35([M+H]+),与理论计算值876.35([M+H]+)相一致,初步证明产物结构。由核磁氢谱图2(b)可知,在磷脂Pyr-SS-PC中,化学位移3.34处信号峰归属于GPC中亲水头胆碱结构-N+(CH3)3的甲基质子峰;化学位移1.24~1.68处信号峰归属于巯基十一烷酸中亚甲基-CH2-质子峰;此外,在7.05~8.42化学位移处出现了典型的Pyr-SS-PC中吡啶环结构质子氢信号峰,特征性证明Pyr-SS-PC磷脂的成功合成。就核磁碳谱图2(c)而言,在54.7(-N+(CH3)3)、28.9(-CH2-)及119.6~149.5(Pyridyl)等处信号峰同样存在,进一步说明产物合成。
综上,HRMS、1H NMR和13C NMR的检测结果表明,已成功制备新型π-π堆积吡啶基双硫Pyr-SS-PC磷脂,且反应转化率良好、纯度高,为下一步纳米脂质体制备及表征提供充足的产物来源。
3.2 DOX/Pyr-SS-PC Lips理化性质
Pyr-SS-PC分子结构中含有疏水烷基长链和亲水磷脂酰胆碱,具两亲性。通过亲疏水的相互作用,在水相体系中能够发生自组装形成纳米脂质体结构。基于此,利用超声波分散法制备DOX/Pyr-SS-PC Lips,即将两亲性Pyr-SS-PC磷脂与其他助磷脂(卵磷脂、胆固醇和DSPE-PEG2000)、药物DOX共混合,真空旋蒸起膜后水化,形成π-π堆积高载药量隐形DOX/Pyr-SS-PC Lips。此外,含有双硫键的Pyr-SS-PC能够赋予脂质体在肿瘤微环境还原响应特性,应激释放负载药物。DSPE-PEG2000嵌入可为DOX/Pyr-SS-PC Lips提供血液长循环作用,减少被内皮网状系统(RES)摄取,避免快速消除。
利用DLS和TEM对制备的DOX/Pyr-SS-PC Lips进行粒径、粒径分布和纳米形貌检测,结果见图3。DOX/Pyr-SS-PC Lips的粒径为160.3 ± 2.6 nm,多分散系数(PDI)值 0.133,Zeta电位-30.8 mV。该结果表明,DOX/Pyr-SS-PC Lips的粒径分布呈现典型的正态分布,PDI值说明脂质体粒径分布均一集中且Zeta 电位处于-40~-20 mV,呈负电性,具有一定的稳定性。据文献报道,肿瘤细胞间距约100~200 nm,正常组织间距50 nm左右。良好的DOX/Pyr-SS-PC Lips粒径分布(100~200 nm)易进入肿瘤组织而非正常组织,实现减毒增效[12]。
从图3(b)可以看到,脂质体呈球形且具有明显的磷脂双分子层结构,视野内大小分布均匀,粒径约为120 nm。此外,TEM图中DOX/Pyr-SS-PC Lips粒径要低于DLS结果,这主要归因于脂质体在进行TEM观察时,处于一种真空干燥的状态,外层亲水性磷脂片段坍塌,造成脂质体皱缩、粒径减小。

图3 DOX/Pyr-SS-PC Lips理化性质表征
3.3 包封率与π-π堆积作用
按照“1.2.2”项下方法制备负载DOX的Pyr-SS-PC功能化脂质体,并以相同药脂比的普通脂质体(DOX/Lecithin Lips)作为对照组,考察Pyr-SS-PC Lips基于药物与磷脂材料之间π-π堆积作用的包封率(EE)和载药量(DLC),结果见表1。

表1 不同药脂比条件下普通DOX/Lecithin Lips与DOX/Pyr-SS-PC Lips的包封率和载药量
如表1所示,随着药脂比由1∶12降至1∶20,两种脂质体的包封率明显增加,但载药量降低。值得注意的是,相比于普通DOX/Lecithin Lips,Pyr-SS-PC磷脂载体与药物DOX分子之间存在较强的π-π堆积作用,使脂质体(药脂比1∶20)的包封率从75.23%提高到85.62%,实现对药物的稳定高效包载。
进一步,利用紫外-可见吸收光谱验证载药纳米脂质体Pyr-SS-PC与DOX之间存在的π-π堆积作用。紫外-可见吸收光谱显示:游离DOX在480 nm处有明显的最大吸收峰,而DOX被负载到Pyr-SS-PC功能化脂质体时,其紫外吸收峰发生偏移至510 nm(图4)。上述结果证明纳米粒中负载DOX与Pyr-SS-PC磷脂之间存在强烈的分子间作用力π-π堆积,引发DOX吸收峰红移。

图4 游离DOX/和DOX/Pyr-SS-PC Lips的紫外光谱
3.4 体外响应释放行为
利用动态透析法考察DOX/Pyr-SS-PC Lips在生理环境下(PBS,pH=7.4)及模拟癌细胞内还原条件下(10 mmol/L GSH,PBS)体外响应释药行为。从图5可以得出,DOX/Pyr-SS-PC Lips在37 ℃、pH=7.4的PBS缓冲溶液中24 h累积释放量< 20%,且前期释放速度较快、后期逐渐变缓,表现出一定的缓释性。然而,在10 mmol/L GSH还原浓度下,脂质体响应释放DOX明显加快,大约87%的DOX在24 h内释放。GSH体外药物释放结果表明DOX/Pyr-SS-PC Lips释药效率显著高于中性PBS条件下释放速率,与预期结果一致。推测其在肿瘤细胞高表达GSH环境下,能够实现还原敏感触发式DOX药物胞内释放,增强药效[13]。
脂质体属于热力学不稳定体系。为了测定DOX/Pyr-SS-PC Lips的体外稳定性,进一步通过脂质体与血清共孵育考察其DOX体外存留率,结果见图6。DOX/Pyr-SS-PC Lips在血浆中,12 h后体外存留率> 90%,说明可以较稳定存在。这可能是由于载体Pyr-SS-PC与药物之间存在强烈的分子间π-π堆积作用,结构更紧密,不利于药物向外自由扩散,提高了稳定性[14]。

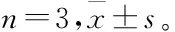
图5 DOX/Pyr-SS-PC Lips在pH=7.4和10 mmol/L GSH条件下的体外释放曲线
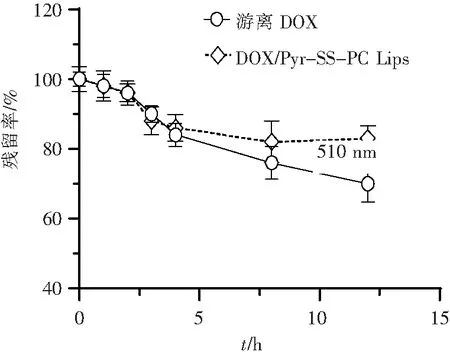
图6 DOX/Pyr-SS-PC Lips的血浆稳定性曲线
3.5 细胞毒性
利用MTT法评估DOX/Pyr-SS-PC Lips在人乳腺癌细胞MCF-7、宫颈癌细胞HeLa和肝癌细胞HepG-2中体外细胞毒性,并计算实验组的半数抑制浓度IC50,结果见表2。游离DOX和DOX/Pyr-SS-PC Lips对MCF-7、HeLa和HepG-2细胞均具有一定的抑制增殖作用,呈现出浓度依赖性特点。然而,DOX/Pyr-SS-PC Lips对于MCF-7、HeLa和HepG-2细胞的IC50均分别略低于游离DOX组IC50。该结果说明在相同浓度下,游离DOX的细胞毒性大于脂质体制剂,主要归因于负载于脂质体中DOX存在水解释放过程,且游离DOX能够通过自由扩散形式进入细胞内发挥活性,而纳米脂质体需要被癌细胞特异性胞吞,摄取过程的差异可能导致DOX/Pyr-SS-PC Lips体外细胞毒性较低。

表2 DOX/Pyr-SS-PC Lips对肿瘤细胞的体外细胞毒性
3.6 体内抗肿瘤活性
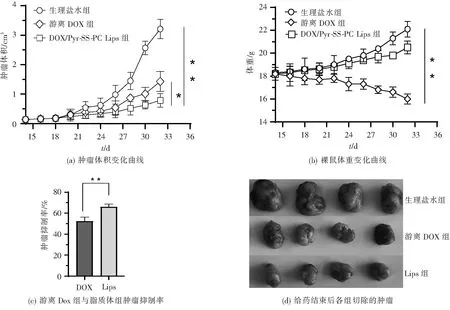
采用荷MCF-7乳腺癌Balb/c裸鼠为动物模型,研究DOX/Pyr-SS-PC Lips的体内抗肿瘤活性。实验结果如图7(a)所示,由于肿瘤的恶性增殖能力,生理盐水组的裸鼠肿瘤块增长迅速,给药结束后体积达到3.23 cm3。在DOX/Pyr-SS-PC Lips给药组肿瘤的增长受到明显的抑制,抑瘤结束后肿瘤仅增长至0.64 cm3,肿瘤抑制率TIR%为65.9%(图7(c)、(d)),且明显高于游离DOX给药组(TIR% 52.31%)。此外,给药脂质体组和生理盐水组的荷瘤裸鼠体重增长速率保持一致,无明显的毒副作用(图7(b))。以上结果说明Pyr-SS-PC Lips能有效负载DOX药物,在动物水平上仍具有良好的抗肿瘤效果,减毒增效。
4 结 论
本研究成功合成了一种新型的π-π堆积的氧化还原型磷脂酰胆碱Pyr-SS-PC。基于π-π堆积作用,该磷脂衍生物能够高效、稳定地负载DOX,粒径为160.3 ± 2.6 nm,且在肿瘤细胞内高GSH条件下响应释药,抑制细胞增殖。尤其突出的是,DOX/Pyr-SS-PC脂质体显著降低游离DOX的体内毒副作用,实现65.9%的MCF-7肿瘤生长抑制率,为乳腺癌的体内治疗奠定基础,并有望成为一种新型的磷脂材料。
